
近日,北京智源大会「AI for Science」分论坛上,清华大学智能产业研究院副研究员周浩以「面向科学发现的生成式人工智能」为主题展开演讲, HyperAI超神经在不违原意的前提下,对周浩教授的深度分享进行了整理汇总。

周浩教授演讲现场
从文本生成到分子设计的跨界探索
本次演讲,周浩教授主要从面向复杂符号的生成式人工智能、微观样本生成所面临的挑战、目前的具体研究内容 3 个方面进行阐述。
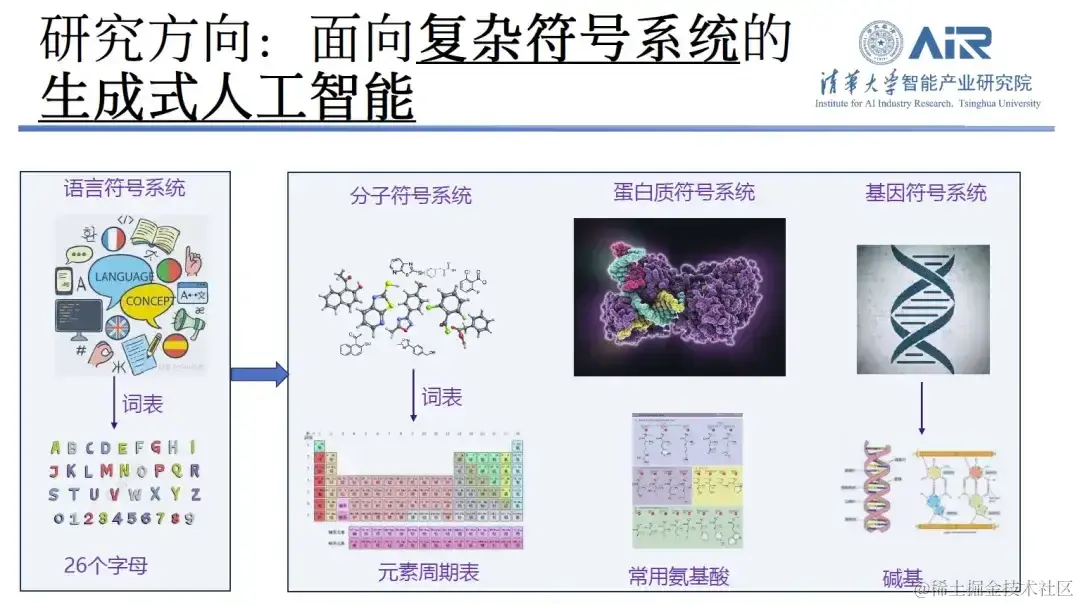
在介绍其相关研究方向时,周浩教授表示,过去 10 年他一直致力于包括文本生成和机器翻译等自然语言的处理。近两年来,其研究重点逐渐从内容创作转向分子生成和蛋白质设计。 在他看来,如果将过去的文本处理工作视为一个复杂的语言符号系统,其中词汇表由 26 个字母构成,那现在的工作就相当于将这 26 个字母扩展到元素周期表、氨基酸、碱基等更广泛的领域。对于这些技术,他的研究团队已经积累了丰富的经验。

从专注于内容创作的人工智能到致力于科学发现的人工智能, 这两者之间存在何种联系?事实上,人工智能可以通过噪声生成完整的图像,许多北美的研究团队也早已利用相似的方法来进行蛋白质设计。通过在空间中随机排列蛋白质的氨基酸,再经过一系列从 0 到 2,000 步的生成设计,就可以设计出外观上相当合理的氨基酸序列。
尽管目前该研究涉及的蛋白质长度还存在一定限制,但近期的研究成果已经显著扩展了这些限制,也暗含了该项技术的巨大潜力,这或许是周浩教授选择该领域的重要原因。
AI 从业者进行 Science 研究时遇到的多重挑战
随后,周浩教授向大家分享了从计算机科学或 AI 领域从业者的角度出发,探索科学领域的人工智能 (AI for Science) 主要面临的 3 大挑战。

第一,分子数据的特异性。 一般而言,文本和符号在处理时是离散的,图像则是 0 到 1 之间的连续信号,但分子数据既包含离散元素,又包含连续元素。
例如,在计算机中存储分子时,研究人员通常将其表示为原子坐标、原子类型,其中原子坐标是连续的,而原子类型是离散的,这形成了一种多模态数据,处理时难度较大。此外,分子还具有几何约束,如旋转、平移的不变性,这在文本或图像处理中并不常见。
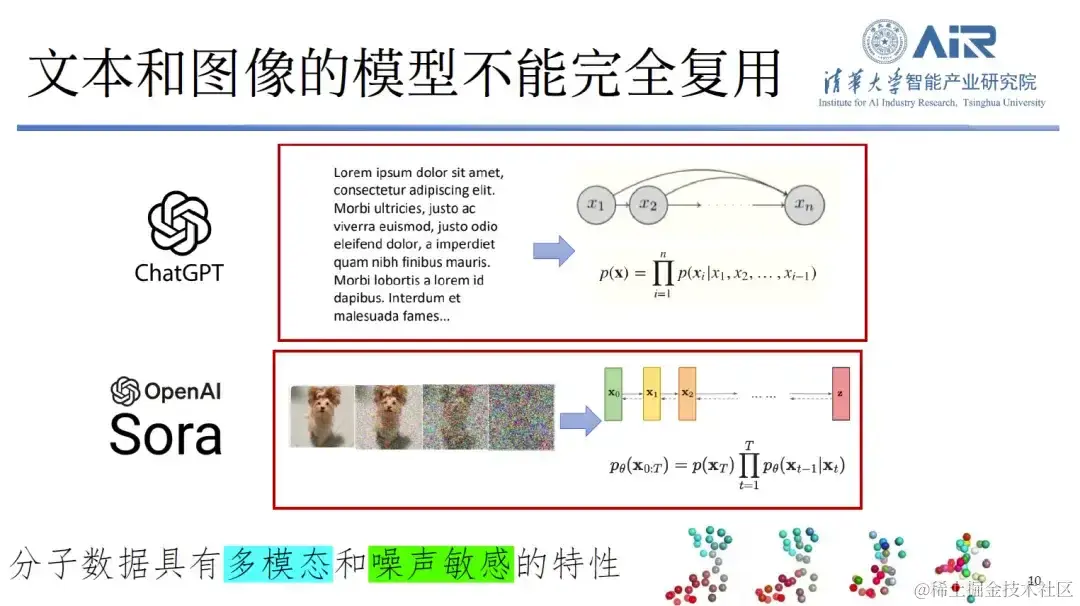
第二,文本和图像的模型在蛋白质领域并不能完全复用。 分子数据不仅具有多模态特性,还对噪声极其敏感。例如,在一张狗的图片上加入噪声,人们仍然能够识别出这是一张狗的图片。但是,如果在分子数据上加入即使很微小的噪声,也可能导致人们无法识别分子的身份,造成大量信息丢失。因此,传统的处理方法并不完全适用于这种新的数据类型。
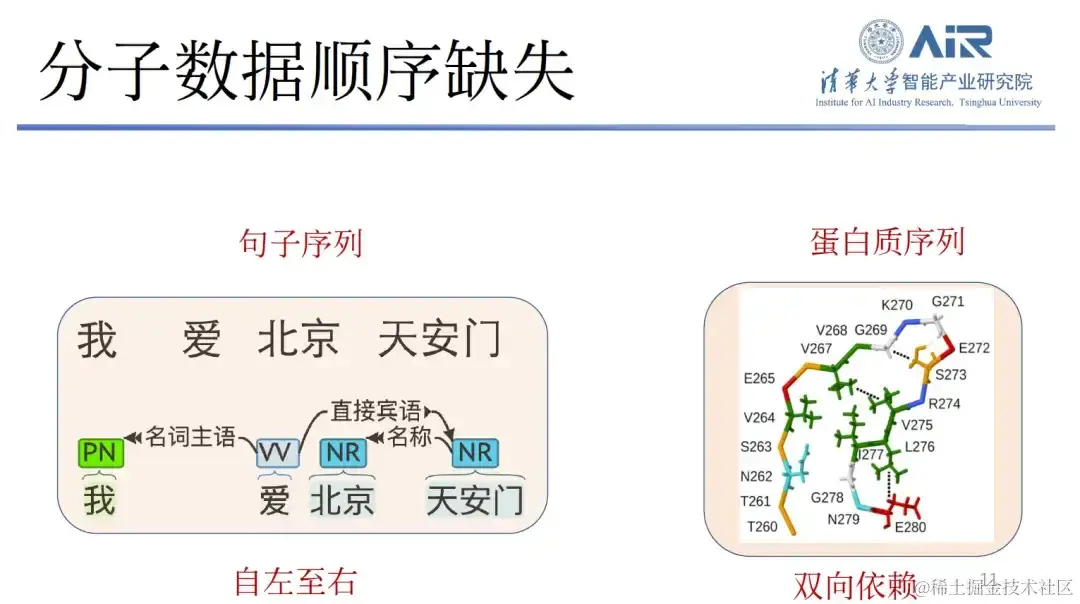
第三,分子数据顺序缺失。 文本对自左至右的依赖非常少,所以它可以通过 GPT 自左至右生成新的文本。但蛋白质的双向依赖性非常强,其前后左右顺序又不易确定,如果直接使用文本或图像模型来生成分子结构,将面临极大的困难。
为了应对上述挑战,周浩教授团队在数据结构、生成算法以及基座构建等方面进行了深入研究。
从数据结构出发,找到本征的数据刻画空间
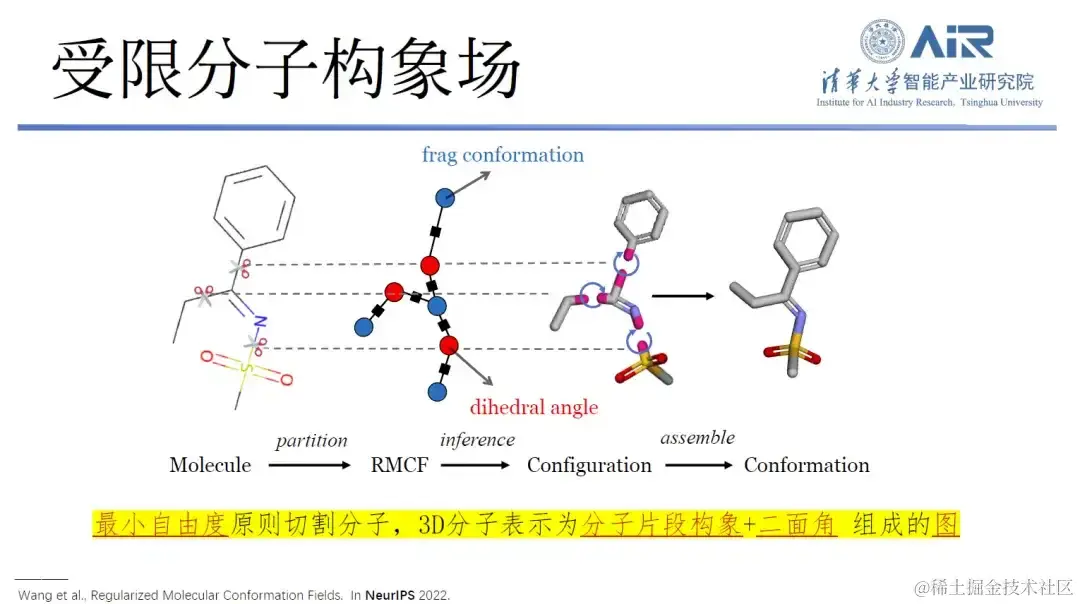
仅保留二面角自由度,重构分子 3D 结构表示
「如何确定分子或目标数据结构的本征空间,是计算机人必须要解决的问题。」周浩教授表示,分子的三维结构表示非常重要,可谓是结构即功能。过去,研究人员主要通过记录原子的坐标、类型构建分子模型,进而获取所需信息。然而,分子的结构很大,又包含大量的冗余信息,如果用过去的方式来建模,从计算机科学的角度来看,这并不是在分子的本征空间中进行观察。

实际上,通过分析分子的键长、键角和二面角就会发现,分子键长、键角的峰值较少,自由度有限,而二面角则有较多的自由度。因此,周浩教授团队设计了一种新方法,即保留二面角自由度的同时,移除其他冗余自由度。
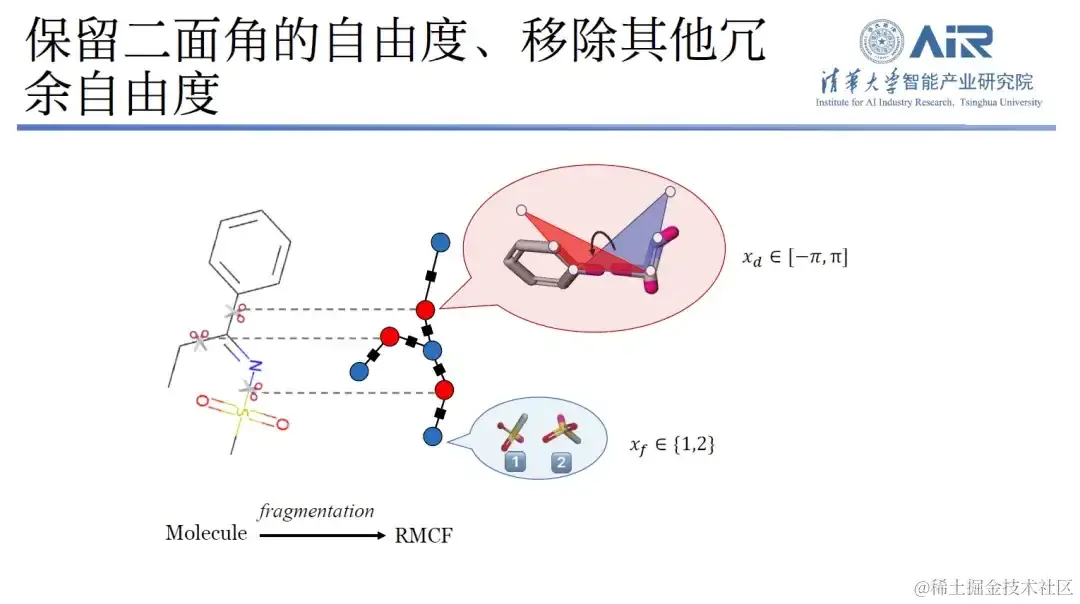
具体来说,该研究可将三维结构转化为二维表示,并通过分子碎片化处理,使得每个分子内部的自由度最小化,而 fragment 之间的自由度最大化,利用动态规划技术,轻松解决 min-max 问题,之后再用算法将所有分子切割成目标数据结构。
论文题目: Regularized Molecular Conformation Fields
论文链接: https://neurips.cc/virtual/2022/poster/53277
「有了这种新的数据结构,如果未来需要进行分子生成,相关研究将会以极少的数据量来构建分子空间,这种思想极其重要!」
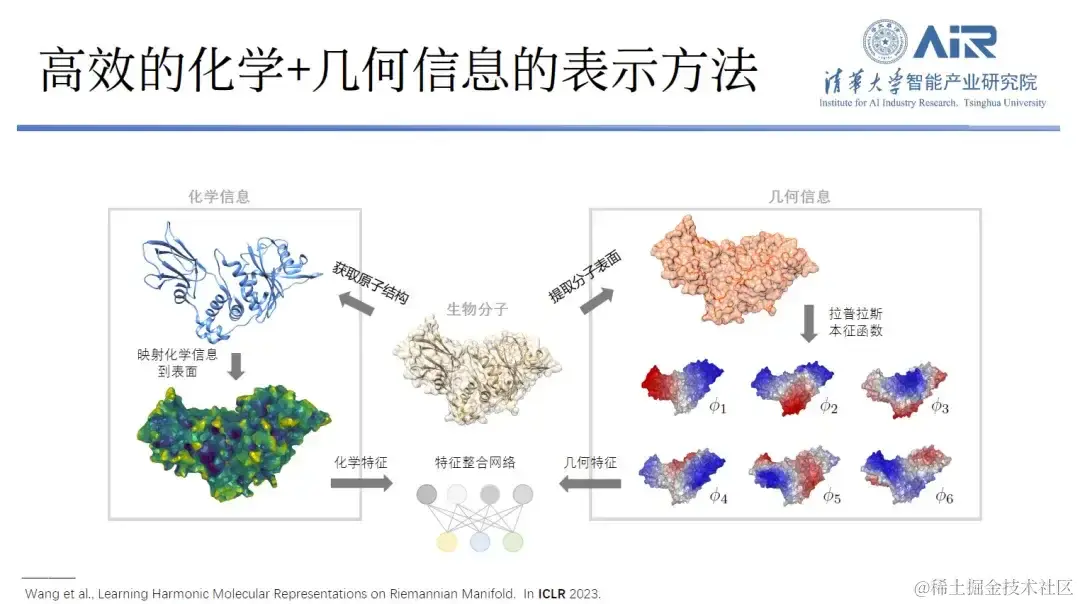
从实空间到谱空间,高效捕捉蛋白质几何、化学信息
除了分子的研究之外,周浩教授团队对蛋白质结构和功能的研究也很感兴趣。
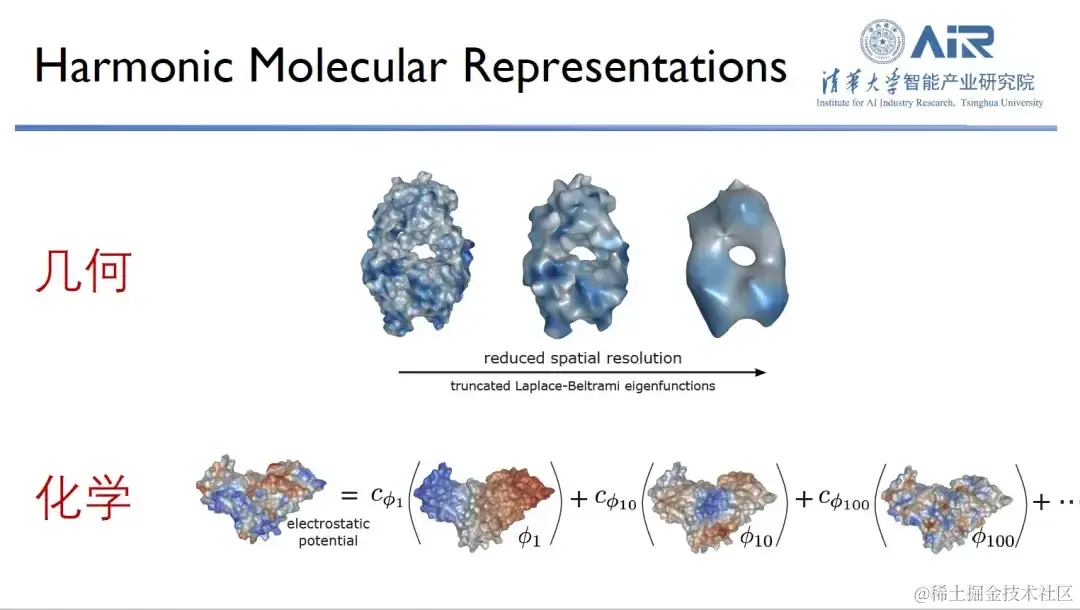
在研究蛋白质时,研究人员通常会从几何信息、化学信息这两个维度来观察。众所周知,蛋白质的形状 (shape) 和表面化学信息对其功能至关重要,只有两者互补,才能表现最佳。
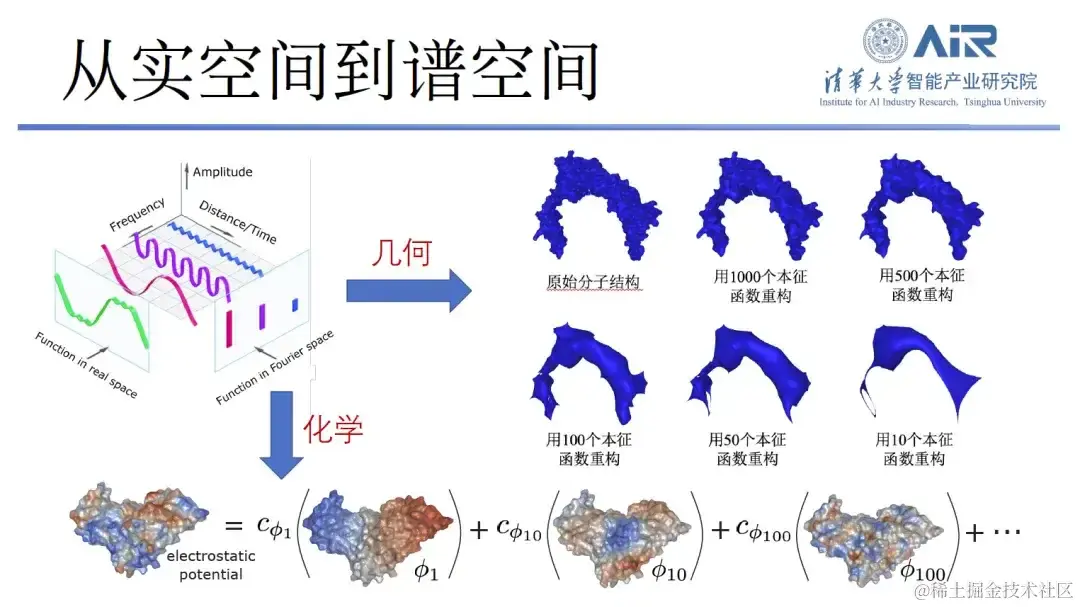
为了高效地表示蛋白质的化学和几何信息, 周浩教授团队将蛋白质从实空间 Transform 到谱空间,再用本征函数来表示蛋白质。例如,使用 10 个本征函数捕捉蛋白质的低频信息,从而解析出它的大致轮廓。另外,越多的本征函数就能捕捉越多的高频信息,通过使用 1,000 个本征函数,就会捕捉到几乎所有的蛋白质信息。
论文题目: Learning Harmonic Molecular Representations on Riemannian Manifold
论文链接: https://iclr.cc/virtual/2023/poster/10900
「以上方法的优势在于,它不仅能够复制蛋白质的几何信息,还能复制其化学信息。」每个本征函数可被视为一个新的空间,蛋白质表面的化学信息可映射到这个本征空间中,在同一个空间既表达几何信息、又表达化学信息,复杂的实空间问题就转换成了简单的谱空间问题。
从生成算法出发,设计适配分子的生成模型
尽管找到了最紧凑、本征的分子和蛋白质空间,但在成功识别这些空间之后,所面临的下一个问题就是:如何利用生成式人工智能有效得到目标分子。
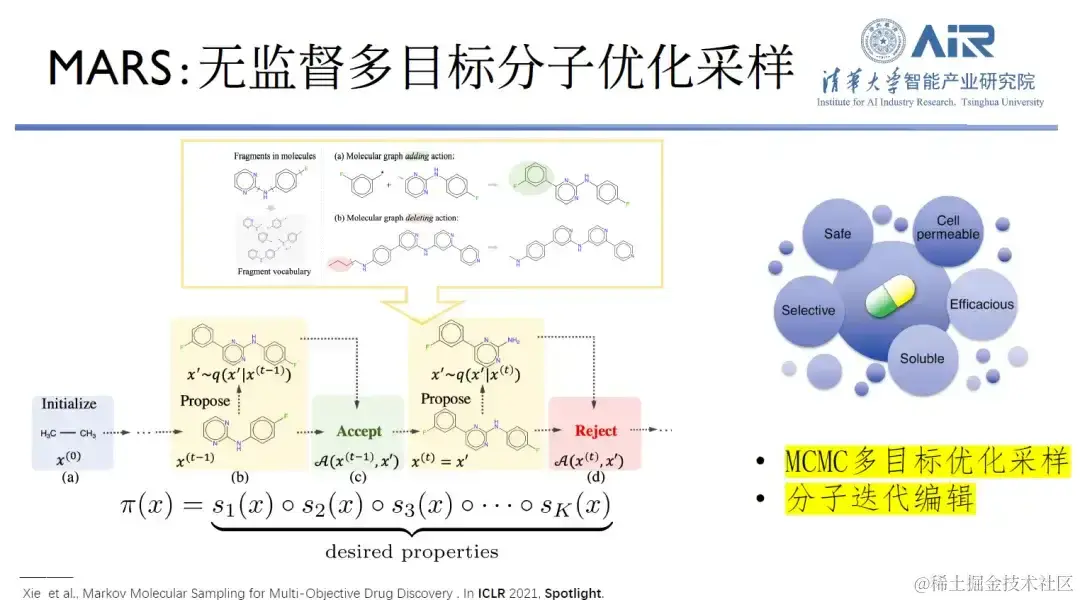
论文题目: MARS: Markov Molecular Sampling for Multi-objective Drug Discovery
论文链接: https://iclr.cc/virtual/2021/poster/3352
为了找到最适配的分子生成模型, 周浩教授团队开发了一种名为 MARS 的模型,该模型采用无监督的多目标分子优化采样来做 2D 的分子设计,其分子设计过程中需要满足多个设计目标,这是一个在复杂高维空间中进行采样的问题。采用马尔可夫链蒙特卡洛 (MCMC) 框架来编辑分子,如果满足细致平衡条件,就能生成任意的目标分子。
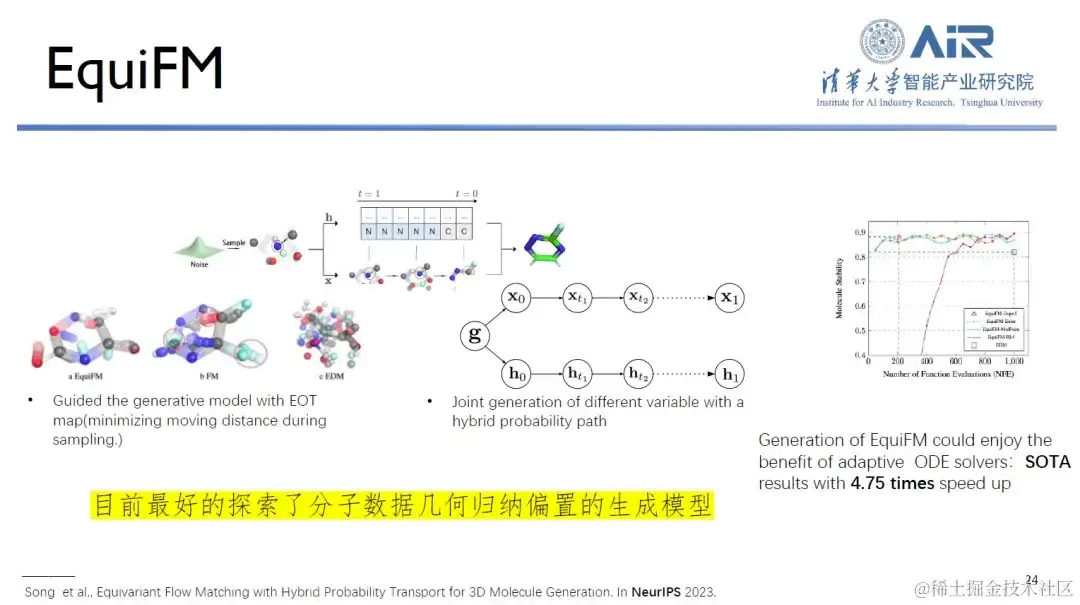
论文题目: Equivariant Flow Matching with Hybrid Probability Transport
论文链接: https://neurips.cc/virtual/2023/poster/70795
同时,周浩教授团队提出的 EquiFM 是目前在探索分子数据几何归纳偏置方面表现最好的生成模型,它在多个分子生成基准测试中都能获得很好的性能,平均采样速度提高了 4.75 倍。
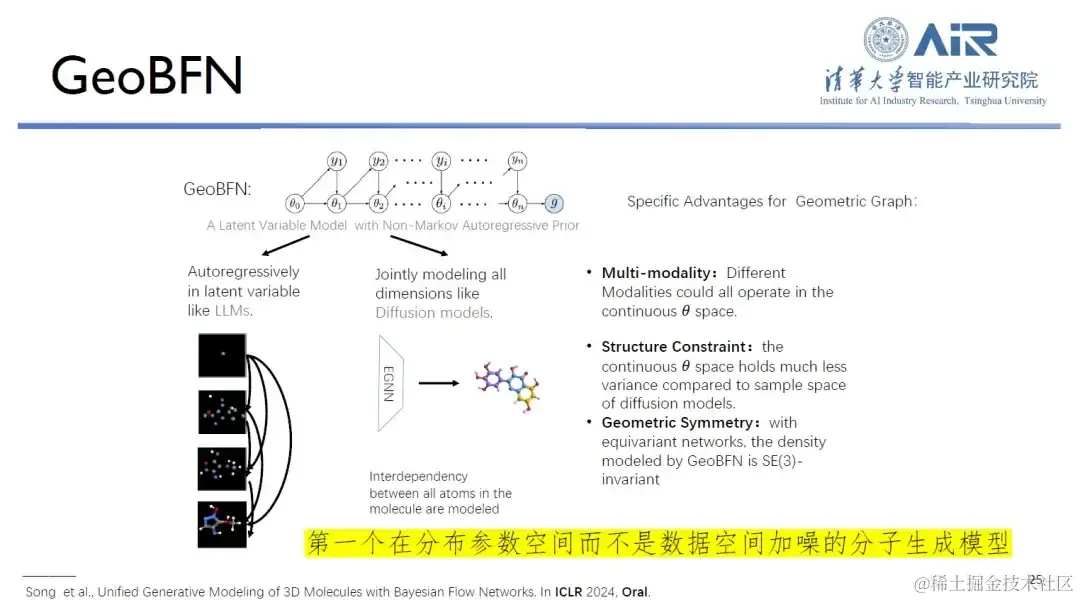
论文题目: Unified Generative Modeling of 3D Molecules via Bayesian Flow Networks
论文链接: https://iclr.cc/virtual/2024/oral/19764
此外,GeoBFN 分子生成模型的核心是将数据空间中的所有分子数据转换到高斯均值方差空间,从而生成具有高合法性和接近真实分布的分子。对此,周浩教授表示:「这是目前最适合分子的深度生成模型,存在极大的发展潜力。」
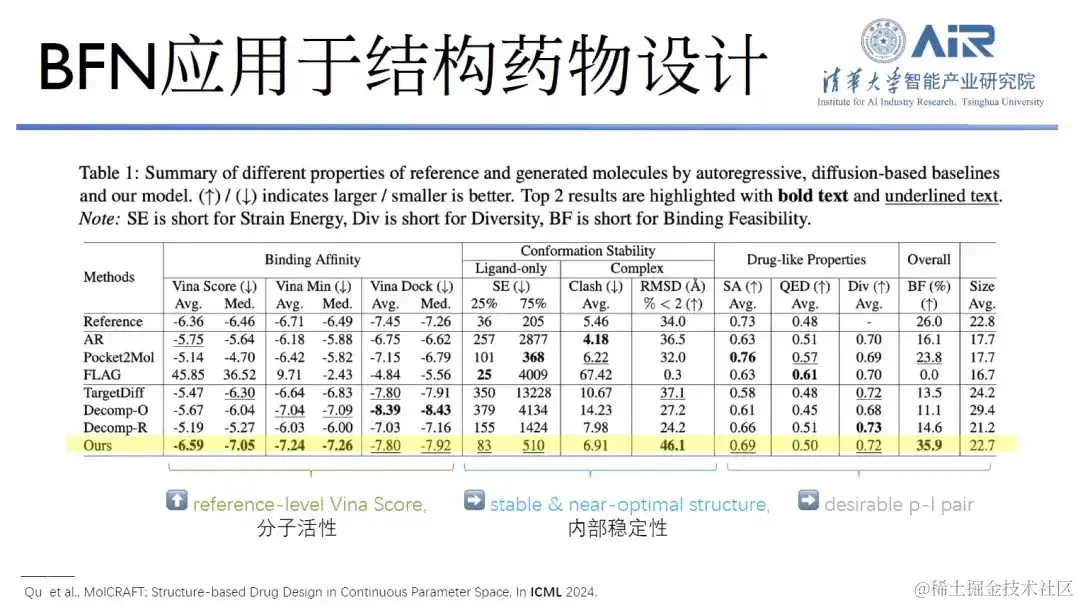
论文题目: MolCRAFT: Structure-Based Drug Design in Continuous Parameter Space
论文链接: https://icml.cc/virtual/2024/poster/34336
除了这些工作,周浩教授团队还曾在国际机器学习会议 (ICML) 上发表了一篇论文,探讨将 GeoBFN 应用于结构药物设计的可能性。研究结果表明,使用该模型生成的分子具有非常稳定的构象和良好活性。
从基座构建出发,建立富含广袤数据知识预训练基座
最后,周浩教授向大家分享了如何从基座构建出发,建立富含广袤数据知识预训练基座。
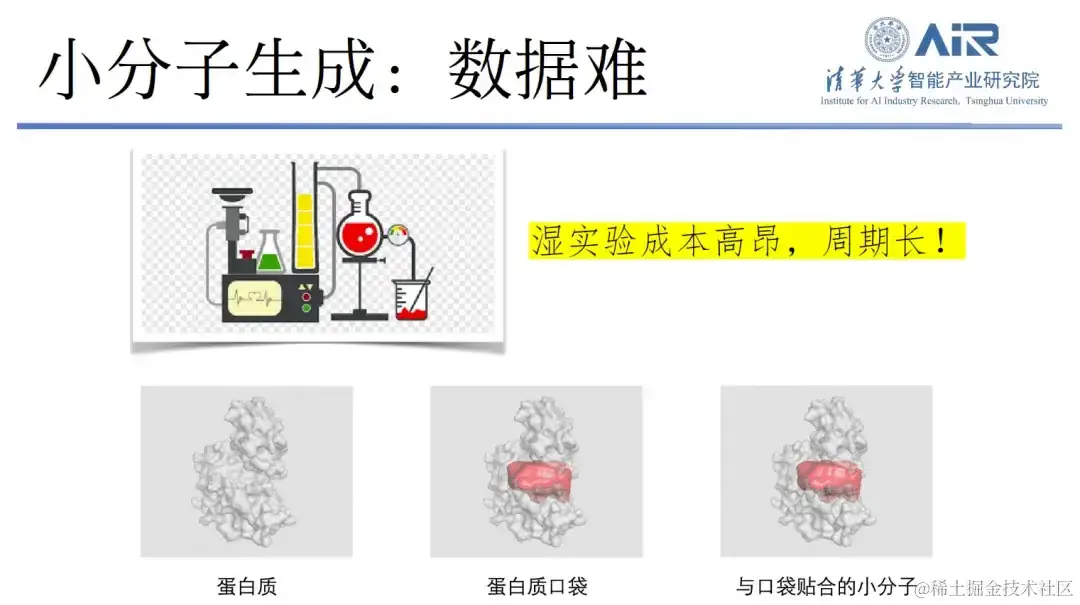
在现有研究中,小分子生成的实验数据十分匮乏,尝试用计算机科学的方法来解决这个问题是一种很重要的思路。
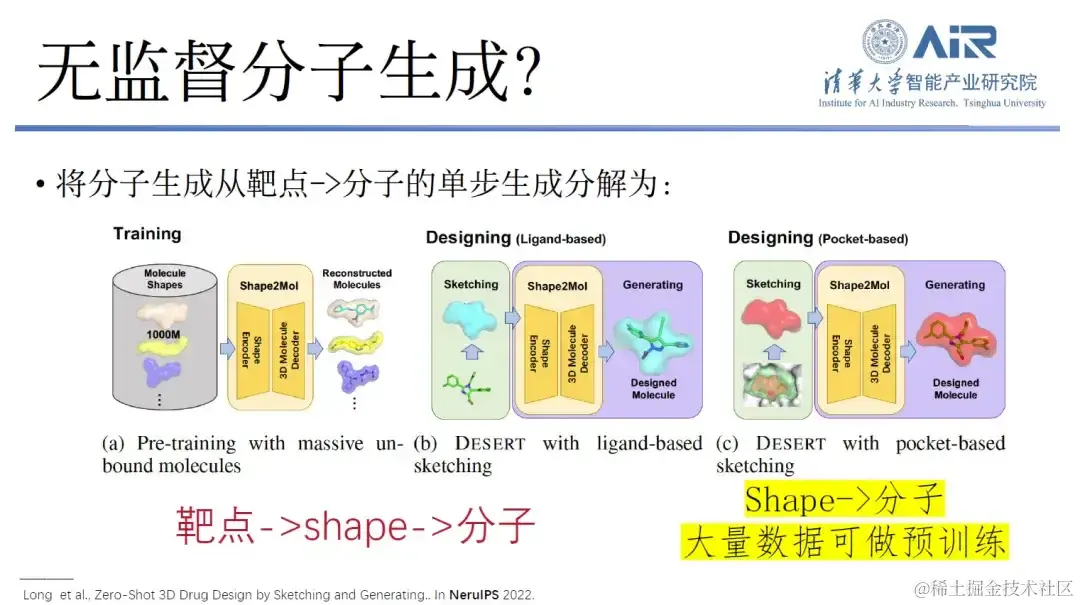
论文题目: Zero-Shot 3D Drug Design by Sketching and Generating
论文链接: https://neurips.cc/virtual/2022/poster/54457
对此,周浩教授团队提出了一个新的想法,即将分子生成从靶点到分子的单步生成分解,变成从靶点到 shape,再从 shape 到分子的过程。 事实上,虽然从靶点直接到分子的数据量很少,但从 shape 到分子的数据量却非常多,这些数据足够从靶点采集各种各样的 shape,再做从 shape 到分子的超大规模预训练模型。最后很快实现从靶点到分子,甚至实现无监督或者少监督的药物分子设计。
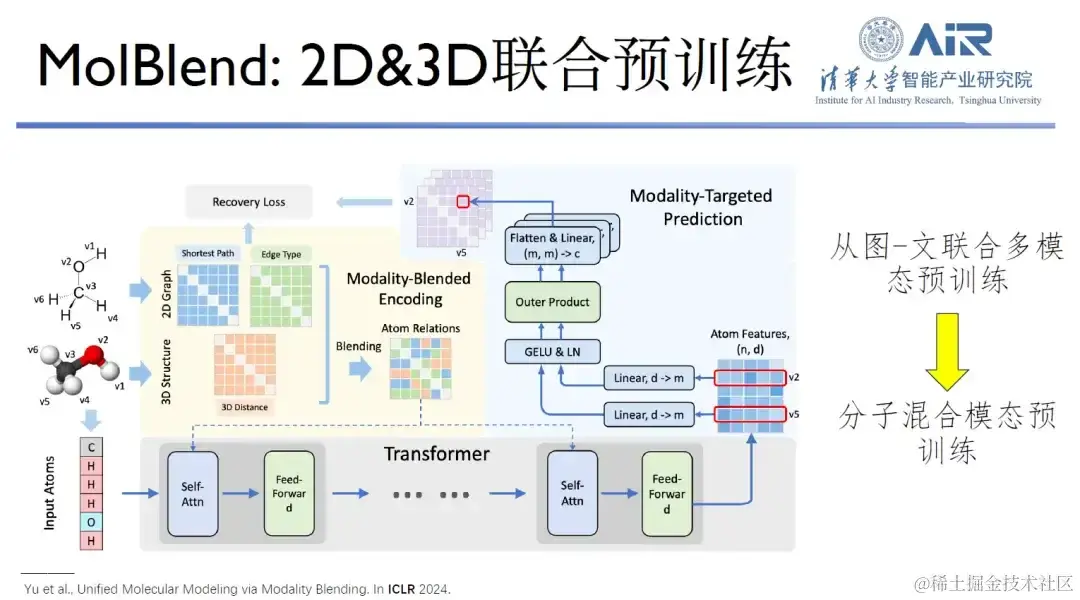
论文题目: Multimodal Molecular Pretraining via Modality Blending
论文链接: https://iclr.cc/virtual/2024/poster/17824
此外,他们提出的 MolBlend 模型,实现了二维和三维分子的联合预训练,这是典型从图文预训练到分子预训练的拓展案例。
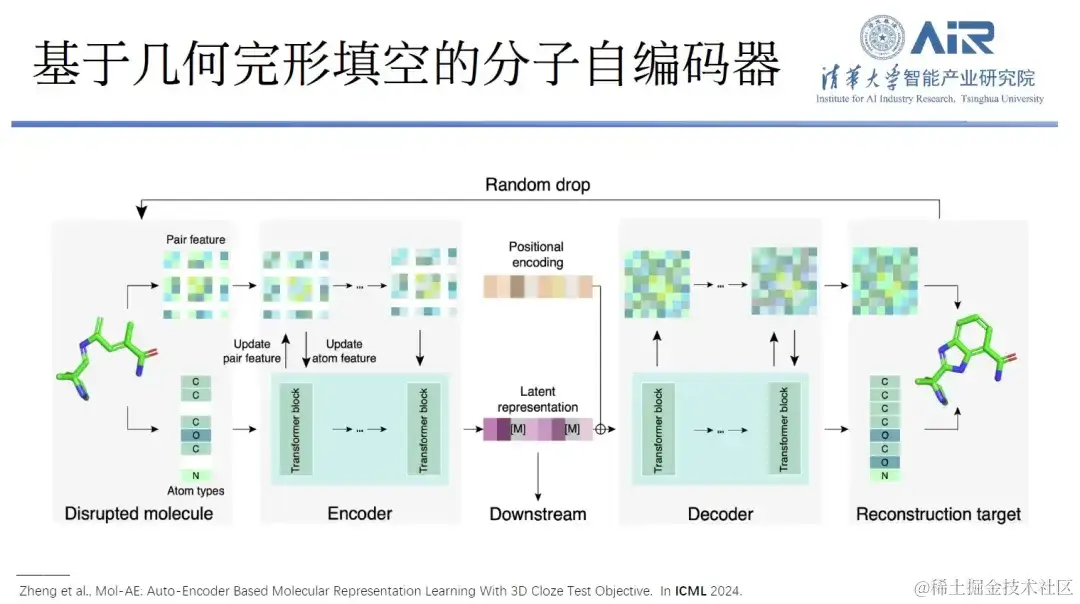
论文题目: Mol-AE: Auto-Encoder Based Molecular Representation Learning With 3D Cloze Test Objective
论文链接: https://icml.cc/virtual/2024/poster/33340
另外,他们还提出了一种基于几何完形填空的分子自编码器 Mol-AE, 和 3D Cloze Test 的新训练目标,所提模型能够更好地学习真实分子结构中的原子空间关系,与目前最先进的三维分子建模方法相比,Mol-AE 实现了较大的性能提升。

蛋白质的通用预训练研究也是他们选择的一个方向。据了解,目前蛋白质的通用预训练主要分为三大类:DeepMind Alphafold 系列、David Baker 的 RoseTTAFold 系列,以及 Meta ESM 系列,周浩教授团队目前开发了其中的 ESM-AA 模型。
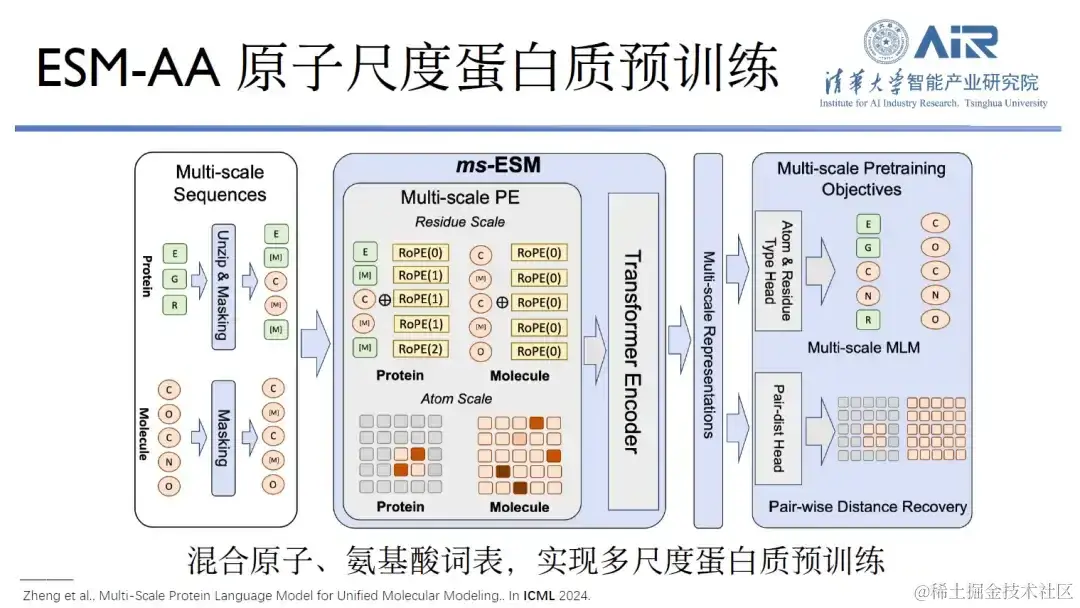
论文题目: Multi-Scale Protein Language Model for Unified Molecular Modeling
论文链接: https://icml.cc/virtual/2024/poster/35119
这是因为,从 Alphafold2 到 Alphafold3 的升级已经构建了全原子的基座,RoseTTAFold 系列同样如此,只有 ESM 系列还没有做全原子的基座。自去年 9 月份以来,周浩教授团队一直在进行这项工作,结合原子和氨基酸词汇表,可多尺度的实现蛋白质训练,在蛋白质和小分子联合任务中,ESM-AA 的表现优于单独预训练基座,如 ESM、其他蛋白质预训练或小分子预训练基座。
这项预训练基座在 Twitter 上也获得了广泛好评。作为序列基座的代表,ESM-AA 后续将与结构基座的代表 RoseTTAFold 和 Alphafold3 竞争,「我想,这也是我们未来的目标。」周浩教授表示。
关于周浩教授
周浩,1990 年生,博士,清华大学副研究员。研究方向是面向复杂符号系统的生成式人工智能,主要的应用包括超大规模语言模型,分子生成,蛋白质设计,新材料发现等。

曾任字节跳动研究科学家和副总监,领导搭建了字节跳动的文本生成中台和 AI 辅助药物设计两个方向的研发团队,研发产品应用于全球 20 余个国家,用户规模超过 10 亿。他长期担任 ICML、NeurIPS,ICLR,ACL 等人工智能顶级会议的领域主席,在人工智能重要国际会议上发表论文 80 余篇。获 2019 年度中国人工智能学会优秀博士论文奖、自然语言处理领域顶级国际会议 ACL 2021 最佳论文奖 (1/3350) 、2021 年度中国计算机学会 NLPCC 青年新锐学者奖等荣誉。