
阅读量:0
零基础入门转录组数据分析——加权基因共表达网络分析(WGCNA,Weighted correlation network analysis)
目录
您首先需要了解本贴是完全免费按实际案例分享基础知识和全部代码,希望能帮助到初学的各位更快入门,但是 尊重创作和知识才会有不断高质量的内容输出 ,如果阅读到最后觉得本贴确实对自己有帮助,希望广大学习者能够花点自己的小钱支持一下作者创作(条件允许的话一杯奶茶钱即可),感谢大家的支持~~~~~~ ^_^ !!!
注:当然这个并不是强制的哦,大家也可以白嫖~~,只是一点点小的期盼!!!
祝大家能够开心学习,轻松学习,在学习的路上少一些坎坷~~~
1. WGCNA基础知识
1.1 WGCNA是什么?
是一种通过分析基因表达数据来识别基因间相互关联模式的方法,这种方法不是简单地看哪些基因的表达量高或低,而是深入挖掘哪些基因的表达变化是相似的。
1.2 WGCNA的目的是什么?
旨在通过基因表达模式的不同,挖掘出相似表达模式的基因,并将这些基因定义为模块(module)。这些具有相似表达模式的基因很可能是紧密共调控的,功能紧密相关的,或是同一条信号通路或过程的成员,因此对其进行分析具有特定的生理意义。
1.3 模块是干什么用的?
模块是用来与外部特征(如时间点、病理进程、表型性状等)进行关联分析的,从而鉴定出与外部特征相关的基因。
1.4 什么是加权?
加权是指对基因之间的相关性值进行幂次运算 (幂次的值也就是软阈值 )
1.5 为什么要加权或者说进行幂次运算
幂次运算的目的是强化相关系数的变化层次,使得网络中的基因之间的连接服从无尺度网络分布
1.6 什么是无尺度分布网络
关于无尺度分布网络的介绍可以参考这一篇教程【WGCNA学习笔记】无尺度分布和软阈值
举个栗子: 各位的导师都是专业领域内的小牛/大牛,他们就算是这个领域内的hub节点,因为他们跟同领域内的其他老师或多或少都会有联系,以你们导师为中心会出现一种成辐射状的发散形式,如下图所示,研究领域相似的人在一个实验室里,呈现在WGCNA中就是表达相似的基因被纳入到一个模块中。
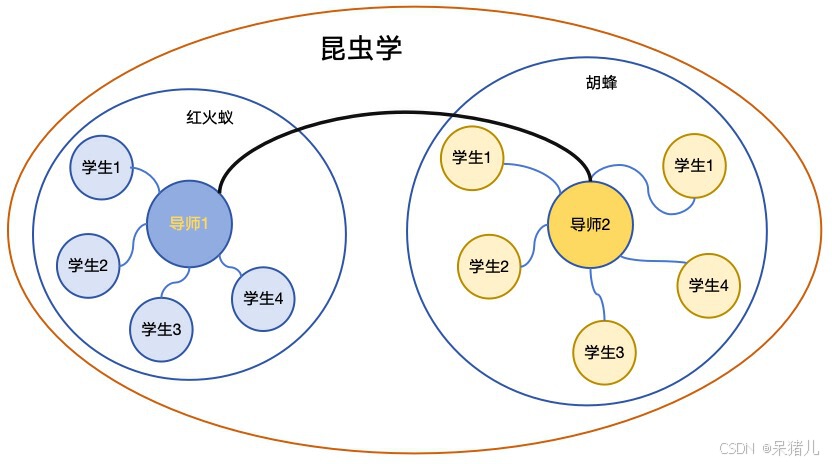
综上所述: WGCNA就是通过加权计算将全部基因中表达模式相似的基因划分到一个模块中,之后对模块与外部特征(如时间点、病理进程、表型性状等)进行相关性分析,与外部特征显著相关的模块被认为是关键模块,里面的基因自然而然的被认为是与该外部特征相关联。
2. WGCNA(Rstudio)——代码实操
本项目以TCGA——肺腺癌为例展开分析 物种:人类(Homo sapiens) R版本:4.2.2 R包:WGCNA,tidyverse,dplyr,ggplot2 废话不多说,代码如下:
2. 1 数据处理
设置工作空间:
rm(list = ls()) # 删除工作空间中所有的对象 setwd('/XX/XX/XX') # 设置工作路径 if(!dir.exists('./07_WGCNA')){ dir.create('./07_WGCNA') } setwd('./07_WGCNA/') 加载包:
library(WGCNA) library(tidyverse) library(dplyr) library(ggplot2) 导入要分析的表达矩阵dat ,并对dat 的列名进行处理(这是因为在读入的时候系统会默认把样本id中的“-”替换成“.”,所以要给替换回去)
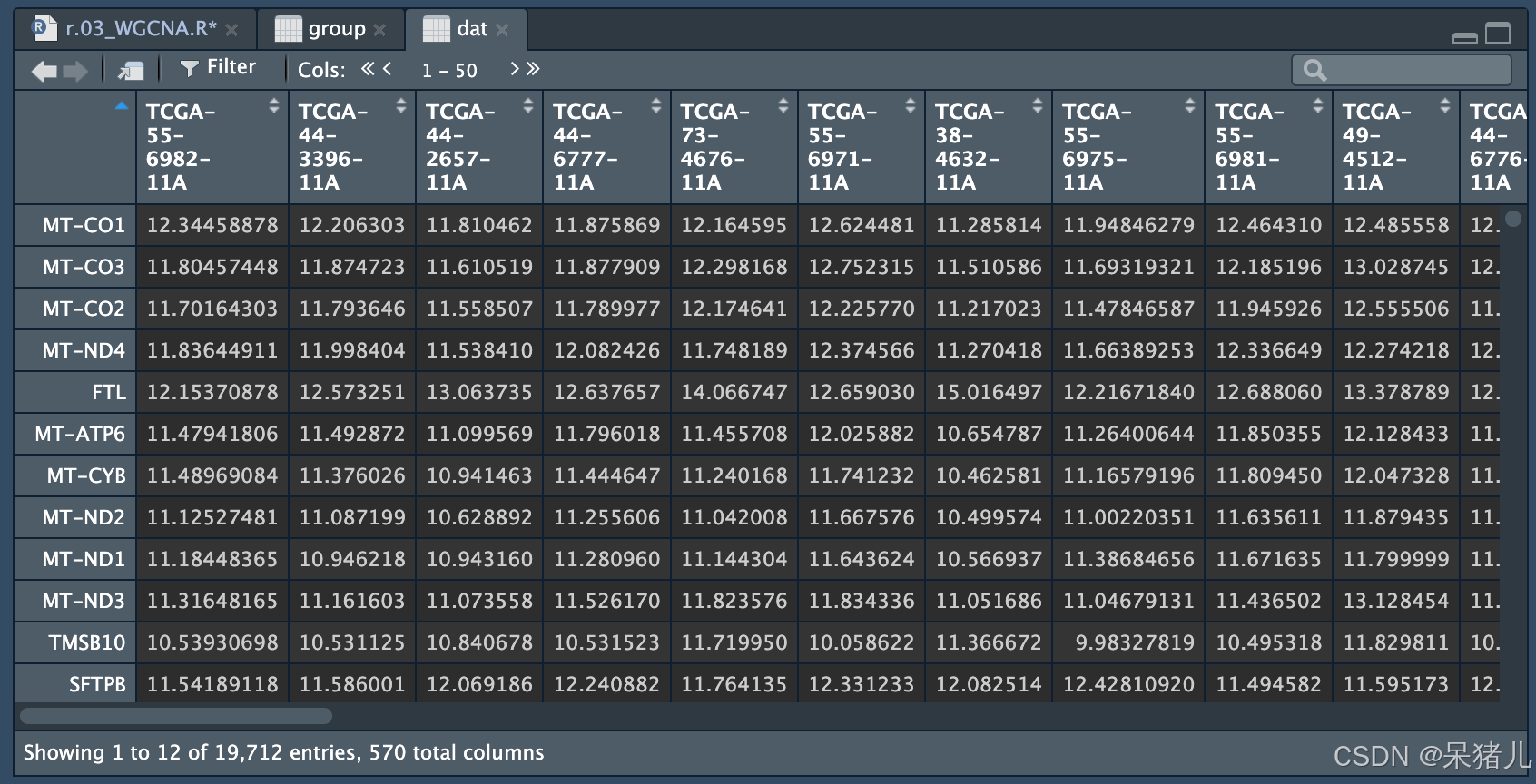
dat <- read.csv('../../00_rawdata/LUAD/data_fpkm.csv', row.names = 1) colnames(dat) <- gsub('.', '-', colnames(dat), fixed = T) dat 如下图所示,行为基因名(symbol),列为样本名
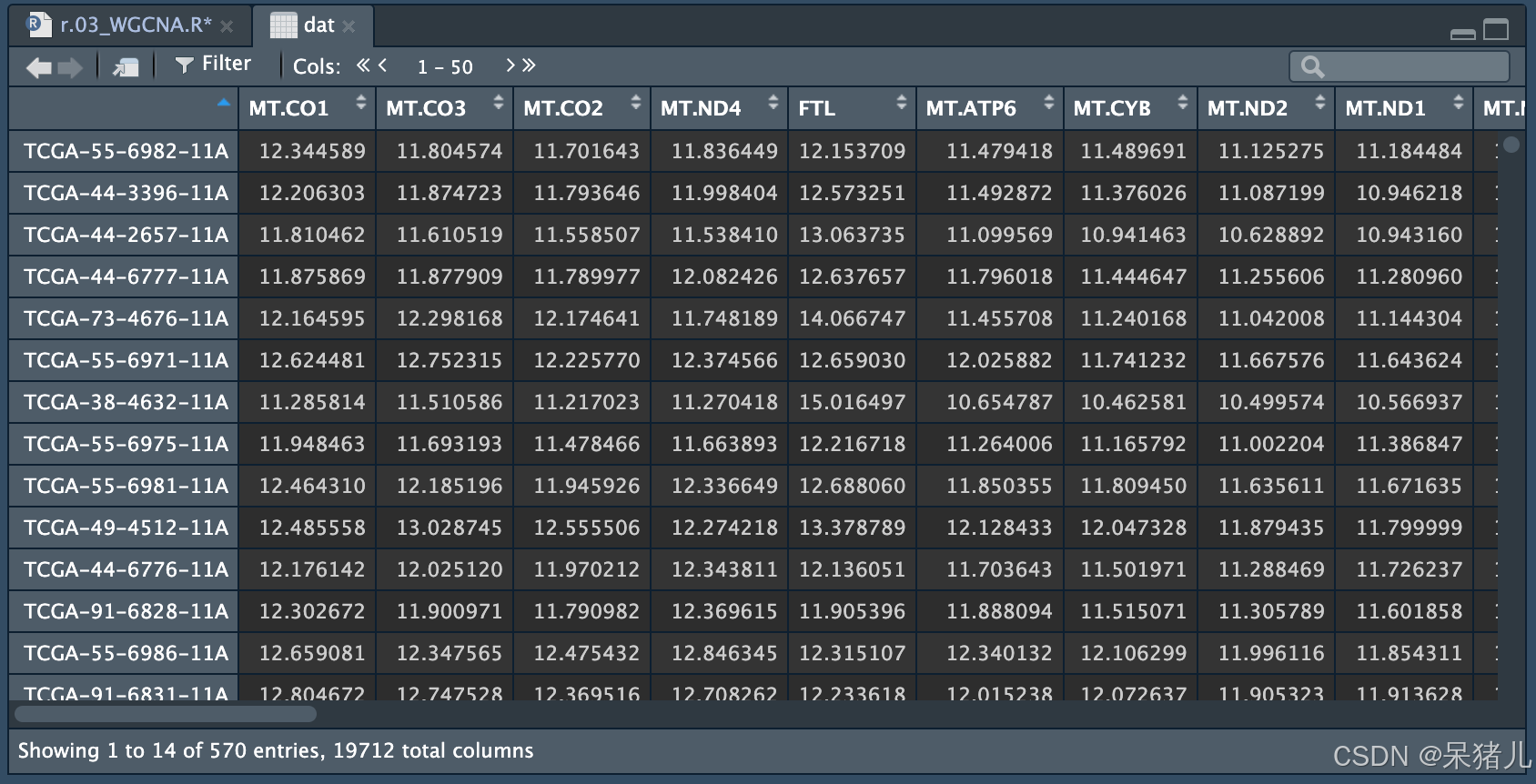
将dat 行和列调转一下,变成行为样本,列为基因的形式
dat <- t(dat) %>% data.frame() 处理后的dat 如下图所示:
之后对dat 做一个检测,检测其中是否存在缺失值,如果存在缺失值要进行处理,否则后面分析会报错。
temp1 <- goodSamplesGenes(dat, verbose = 3) # 对文件做检测,检测是否有缺失值,即NA temp1$allOK # 如果temp1$allOK的结果为TRUE,证明没有缺失值,可以直接下一步 # 如果temp1$allOK的结果为FALSE,证明有缺失值,执行以下代码: if (!temp1$allOK){ # Optionally, print the gene and sample names that were removed: if (sum(!temp1$goodGenes)>0) printFlush(paste("Removing genes:", paste(names(dat)[!temp1$goodGenes], collapse = ", "))); if (sum(!temp1$goodSamples)>0) printFlush(paste("Removing samples:", paste(rownames(dat)[!temp1$goodSamples], collapse = ", "))); # Remove the offending genes and samples from the data: dat = dat[temp1$goodSamples, temp1$goodGenes] } temp1 <- goodSamplesGenes(dat, verbose = 3) # 对文件做检测,检测是否有缺失值,即NA temp1$allOK 处理后就可以开始准备分组信息表——group
注意:分组信息表是为了在绘制样本聚类树的时候添加一个分组条带,并且后面划分出模块之后,要和模块进行相关性分析
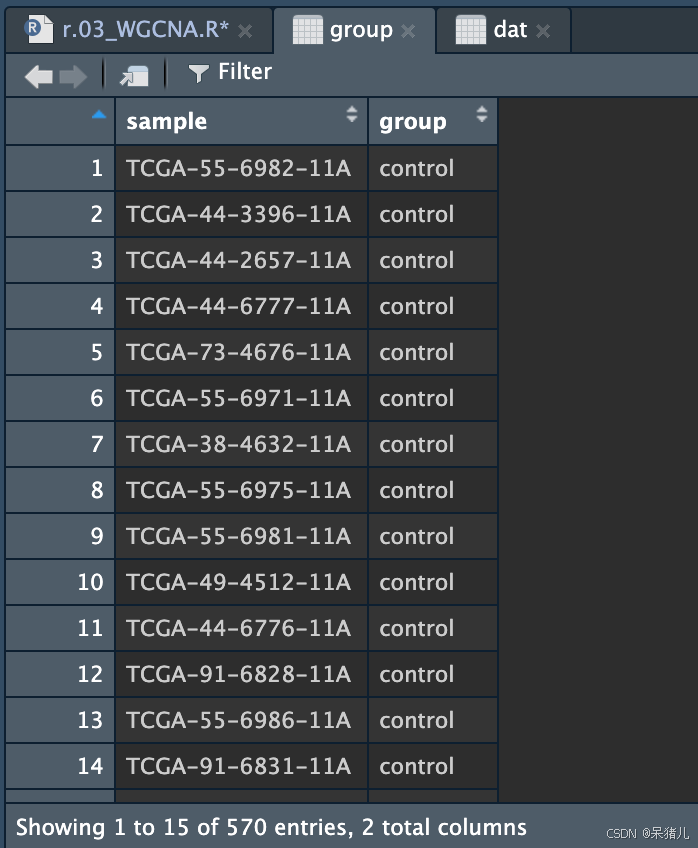
## 分组信息表 group <- read.csv('./data_group.csv', row.names = 1, check.names = F) 导入的分组信息表——group 如下图所示:有两列,第一列是样本对应的id,第二列是样本对应的分组情况(我这里分的是disease组和control组)。
对group 进一步做处理
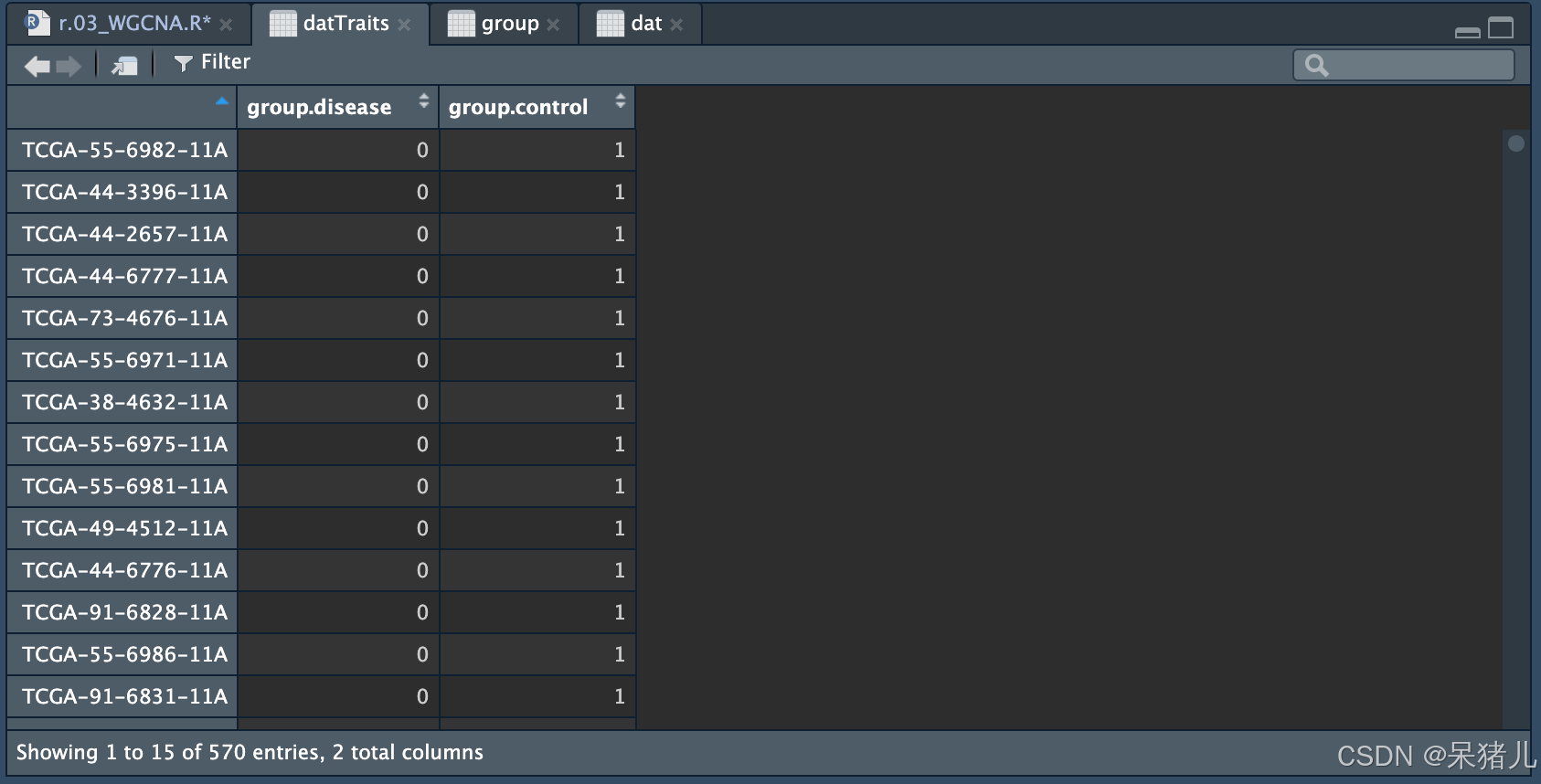
group$disease <- ifelse(group$group == 'disease', 1, 0) group$control <- ifelse(group$group == 'control', 1, 0) group <- dplyr::select(group, -group) group <- plyr::rename(group, c(sample = 'id')) group$id<- lapply(strsplit(group$id, "\\."), function(x) paste0(x, collapse = "-")) group$id datTraits <- data.frame(row.names = group$id, group = group[, 2:3]) 处理后的datTraits 如下图所示,行名为样本名,第一列是疾病组,是疾病的样本赋值为1,不是疾病的样本赋值为0;第二列是对照组,是对照的样本赋值为1,不是对照的样本赋值为0.
之后对datTraits 赋予不同的色号
traitColors <- numbers2colors(datTraits, signed = FALSE) traitColors 如下图所示,里面的#…就是代表不同的颜色,这里面的两列就是上面datTraits 中对应的两列。
2. 2 构建样本聚类树
做样本聚类树(目的:看是否具有离群样本)
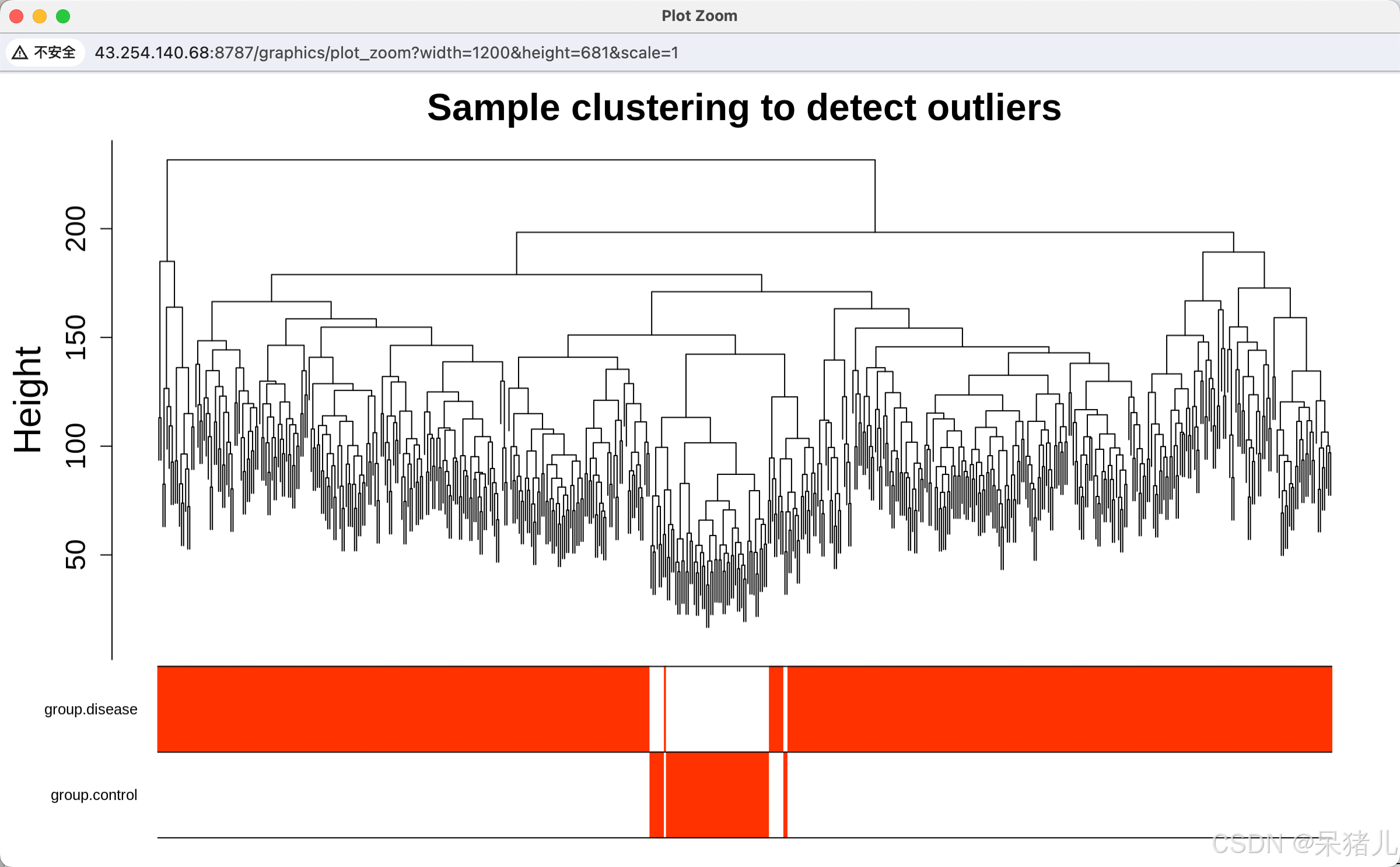
sampleTree = hclust(dist(dat, method = 'euclidean'), method = "complete") # 构建聚类树,看是否有离群样本 通过plotDendroAndColors函数绘图,看一下是否存在离群样本
plotDendroAndColors(sampleTree, traitColors, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 2, # cex.lab指定轴标签的大小 cex.axis = 1.5, # cex.axis指定刻度线标签的大小 cex.main = 2, groupLabels = colnames(datTraits), dendroLabels = FALSE ) 样本聚类树如下图所示,可以看到聚类树中不存在显著离群样本。而下方的那个红色条带就是上一步准备的那个颜色表traitColors
2. 3 计算软阈值
在确认了没有离群样本之后就可以去计算软阈值了,关于软阈值的概念前述已经提及。
datExpr <- dat nGenes = ncol(datExpr) # 列是基因名 nSamples = nrow(datExpr) # 行是样本名 ## 计算软阈值 powers <- c(c(1:10), seq(from = 12, to=30, by=2)) # 设置网络构建参数选择范围,计算无尺度分布拓扑矩阵 sft <- pickSoftThreshold(datExpr, powerVector = powers, verbose = 5) # pickSoftThreshold: will use block size 2284. # pickSoftThreshold: calculating connectivity for given powers... # ..working on genes 1 through 2284 of 19581 # ..working on genes 2285 through 4568 of 19581 # ..working on genes 4569 through 6852 of 19581 # ..working on genes 6853 through 9136 of 19581 # ..working on genes 9137 through 11420 of 19581 # ..working on genes 11421 through 13704 of 19581 # ..working on genes 13705 through 15988 of 19581 # ..working on genes 15989 through 18272 of 19581 # ..working on genes 18273 through 19581 of 19581 计算完软阈值,进行简单可视化,看看软阈值大概多少
par(mfrow = c(1, 2)); # 一张画布上显示两个图 cex1 = 0.9; plot(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3])*sft$fitIndices[, 2], xlab = "Soft Threshold (power)", ylab = "Scale Free Topology Model Fit,signed R^2", type = "n", main = paste("Scale independence")); text(sft$fitIndices[, 1], -sign(sft$fitIndices[, 3])*sft$fitIndices[, 2], labels = powers, cex = cex1, col = "red"); abline(h=0.85,col="red") plot(sft$fitIndices[, 1], sft$fitIndices[, 5], xlab = "Soft Threshold (power)", ylab = "Mean Connectivity", type = "n", main = paste("Mean connectivity")) text(sft$fitIndices[, 1], sft$fitIndices[, 5], labels = powers, cex = cex1, col = "red") 结果如下图所示,通常选择首次越过R^2 = 0.85红线的值作为软阈值(下图中选择6就是比较合适的)
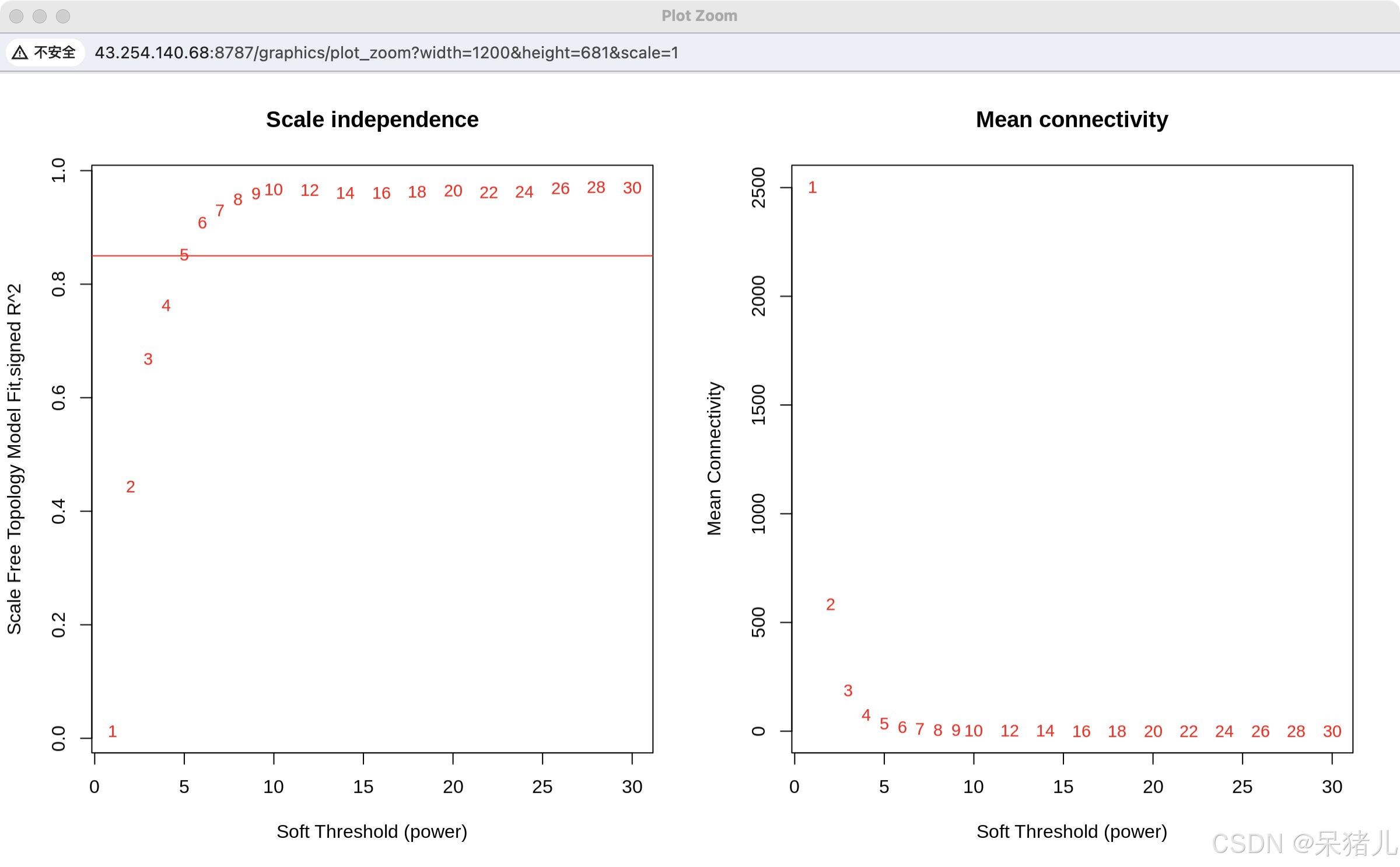
注:同时还要看右侧的纵坐标,必须保证选的值(6)其所对应的平均链接度(Mean connectivity)接近于0
2. 4 一步法构建WGCNA网络
在确认了软阈值,用选定的6带入函数中构建WGCNA网络
一步法构建WGCNA网络需要的东西:
(1)datExpr(表达矩阵,也就是之前的dat)
(2)nGenes(表达矩阵中的基因数目)
(3)power = 6(软阈值,上一步计算的结果)
net <- blockwiseModules(datExpr, power = 6, # 7是刚才picksoftThreshold的软阈值 maxBlockSize = nGenes, # 默认maxBlockSize是5000,表示仅会分析5000个基因 TOMType = "unsigned", # 计算TOM矩阵时,是否考虑正负相关性;默认为"signed",可选"unsigned" minModuleSize = 100, # minModuleSize:模块中最少的基因数,设置为50,即模块中只有大于50个基因才会被统计 reassignThreshold = 0, # deepSplit = 2, mergeCutHeight = 0.3, # mergeCutHeight :模块合并阈值,阈值越大,模块越少(重要) numericLabels = TRUE, # 返回数字而不是颜色作为模块的名字,后面可以再转换为颜色 pamRespectsDendro = FALSE, saveTOMs = TRUE, # 询问是否保存TOM文件 saveTOMFileBase = "gene_TOM", # 保存的TOM文件命名 corType = "pearson", # 计算相关性的方法;可选pearson(默认),bicor。后者更能考虑离群点的影响 loadTOMs = TRUE, verbose = 3) 注:里面的参数就不一一介绍了,具体的可以参考blockwiseModules这个函数的说明书,常用参数后面都有相应的注释
查看聚类出来多少模块,并给模块分别赋予一个颜色(这里一共聚类出13个模块)
table(net$colors) # 结果表示一共可以分为13个模块,第二行是每个模块对应的基因数,由多到少 # 0 1 2 3 4 5 6 7 8 9 10 11 12 13 # 4687 4646 2249 1617 1505 965 948 669 570 402 380 360 307 276 mergedColors <- labels2colors(net$colors) # 给每一个模块分别给个颜色(彩虹色) 2. 5 绘制基因动态剪切树
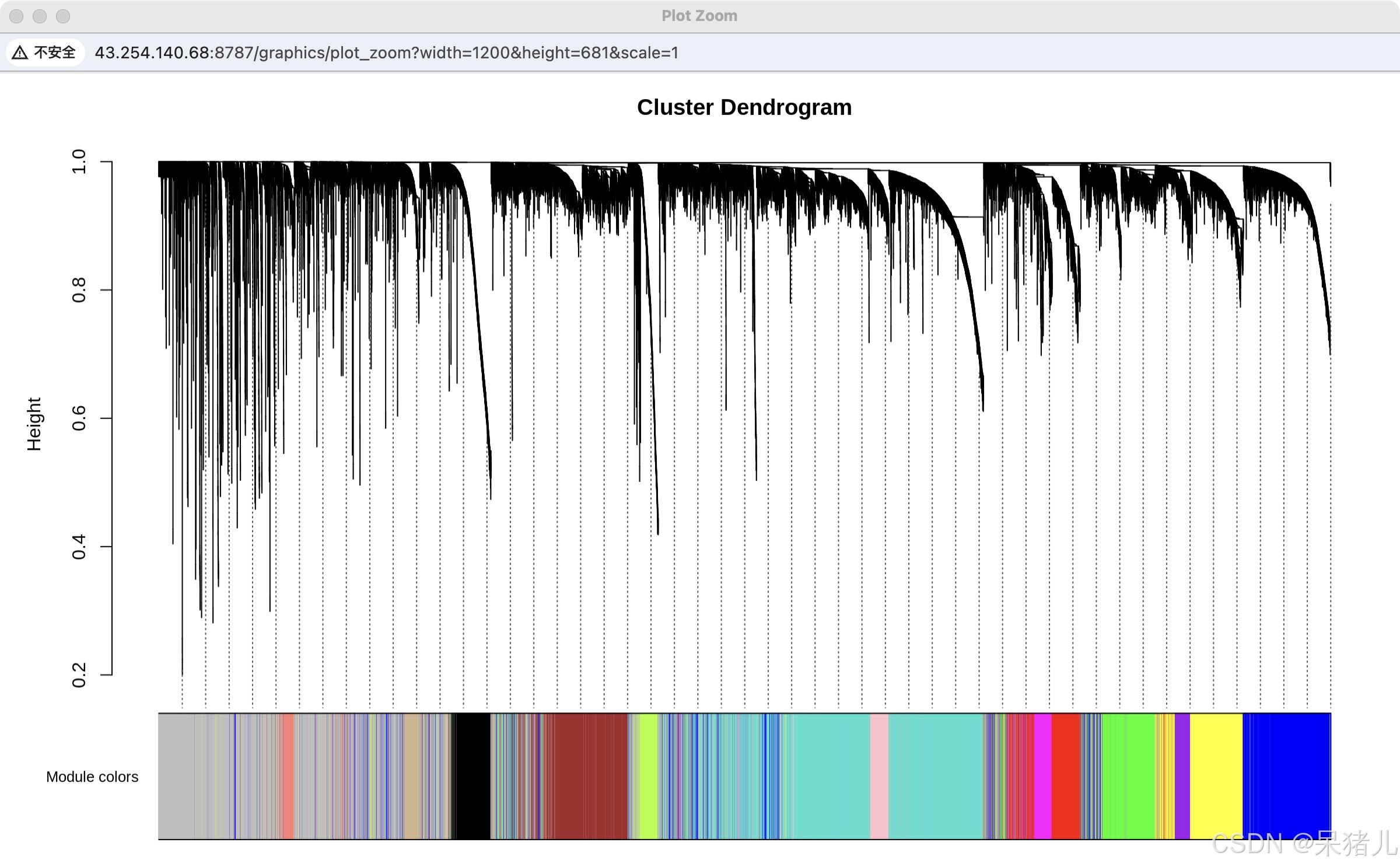
sizeGrWindow(12, 9) plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]], "Module colors", dendroLabels = FALSE, hang = 0.03, addGuide = TRUE, guideHang = 0.05) 结果如下图所示,跟前面样本聚类树很像,但是这里面上方每一个黑线代表基因,表达模式的基因会被划分到一个簇中,每个簇对应下方的一种颜色。
比如:下方蓝色的条带上方对应的基因就是一簇。
2. 6 计算模块与性状相关性(重要)
通过一步法对基因进行聚类后得到不同的模块,那么哪些模块与自己想要研究的性状相关呢?
接下来就要对模块与性状进行相关性分析,首先要计算每个模块对应的ME分数
ME: Module Eigengene,模块特征基因,用于描述和代表整个模块(Module)的基因表达谱,简单来说,ME就是一个能够代表模块整体表达趋势的“分数”或“值”。
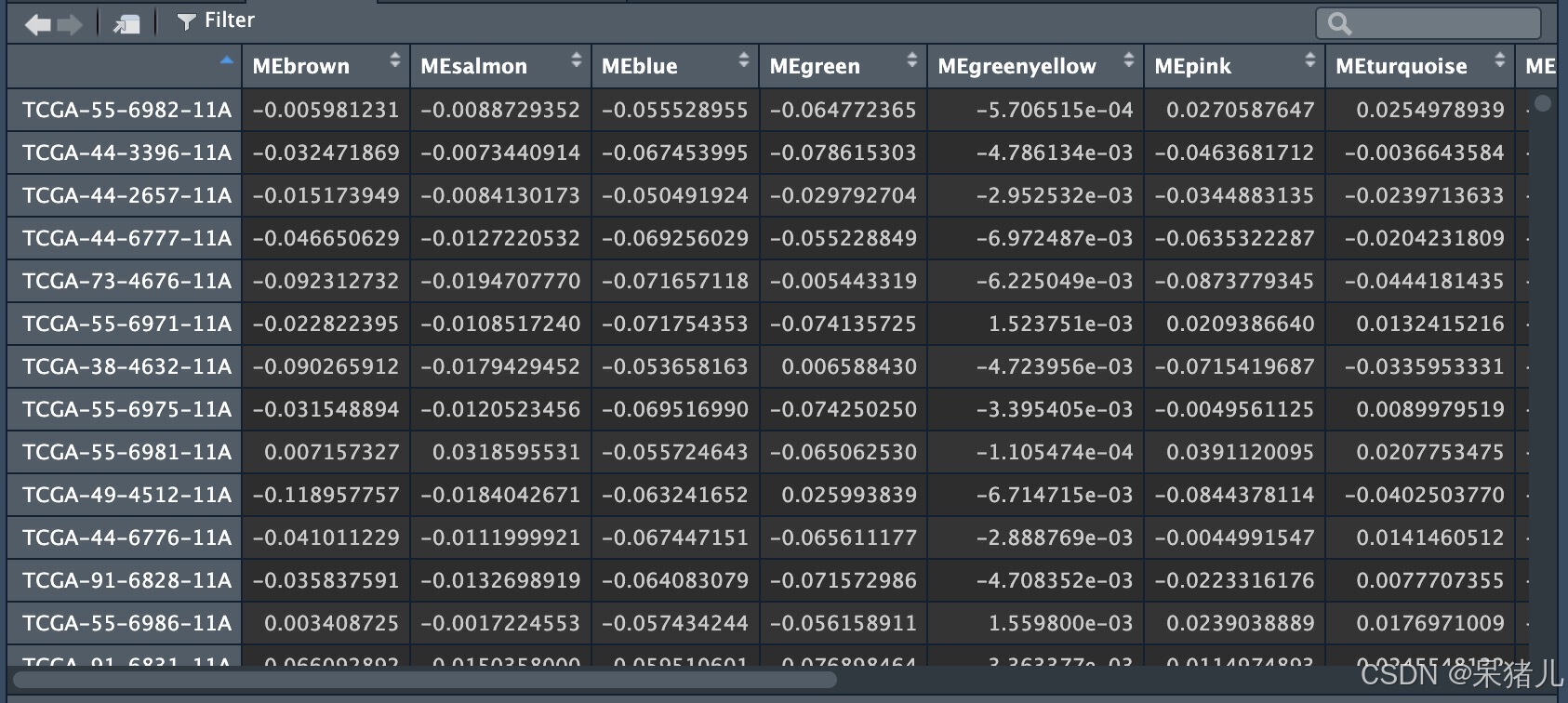
moduleLabels <- net$colors # # 显示了数据集中的每个基因被分配到什么模块中,不同的模块用不同的数字表示 moduleColors <- labels2colors(net$colors) # label_to_colors,将每个模块对应的数字转成颜色 table(moduleLabels) table(moduleColors) ## Recalculate MEs with color labels MEs0 <- moduleEigengenes(datExpr, moduleColors)$eigengenes MEs <- orderMEs(MEs0) MEs 如下图所示,行为样本名,列为模块对应的颜色,表中数值就代表该模块在不同样本中对应的ME分数,
接下来就是要计算这些模块(MEs)与性状(datTraits)之间的相关性
datTraits <- plyr::rename(datTraits, c(group.disease = 'disease', group.control = 'control')) ## 计算基因集和模块之间的相关性 MEs <- dplyr::select(MEs, -MEgrey) ## 剔除灰色模块 moduleTraitCor = cor(MEs, datTraits, use = "p"); moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nSamples) 注:这里要注意下需要剔除灰色模块,因为灰色模块中的基因是一些孤立的基因聚合的模块(实用意义不大)
2. 7 绘制模块与性状相关性热图(重要)
## Will display correlations and their p-values, textMatrix <- paste(signif(moduleTraitCor, 2), "\n(", signif(moduleTraitPvalue, 1), ")", sep = ""); dim(textMatrix) <- dim(moduleTraitCor) ## 绘图保存 par(mar = c(4, 9, 2, 2)) labeledHeatmap(Matrix = moduleTraitCor, xLabels = names(datTraits), yLabels = names(MEs), ySymbols = names(MEs), colorLabels = FALSE, colors = blueWhiteRed(50), textMatrix = textMatrix, setStdMargins = FALSE, cex.text = 0.7, zlim = c(-1,1), main = paste("Module-trait relationships")) 热图结果如下所示:红色的方块表示正相关,蓝色的表示负相关,颜色越深表明相关性系数越大(也就说明模块与性状越相关)
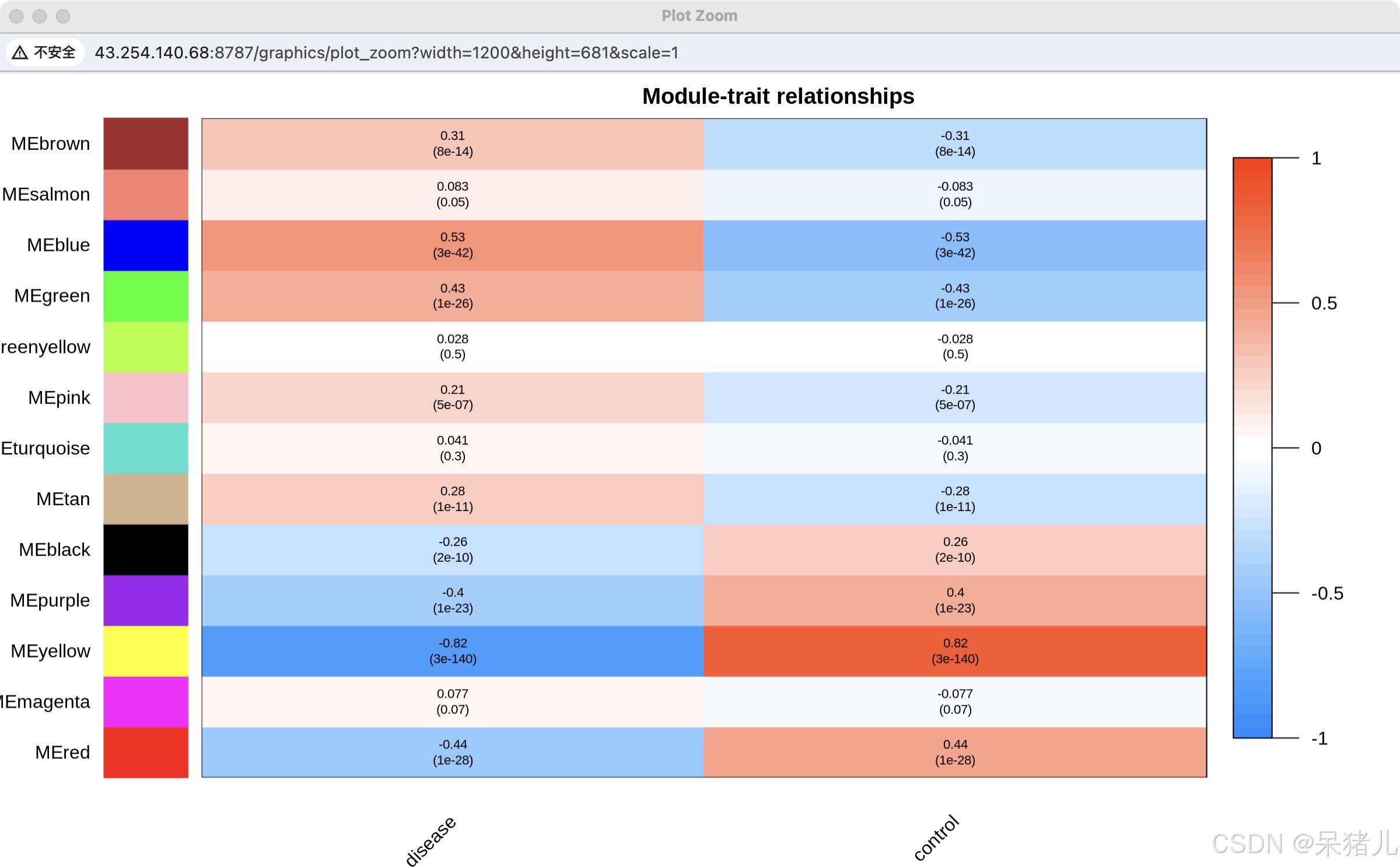
注:细心的朋友可以发现disease和control的相关性系数是一样的,只不过颜色相反,这是正常情况,因为性状分组一般都是二分类(0/1),对于疾病是正相关,那么对于对照也就是负相关。
2. 8 计算关键模块中的GS和MM(非必须)
从上图中我们可以看到黄色模块的相关性系数在所有模块中是最大的,就认为该模块与我们的疾病(肺腺癌)最相关,那么里面的基因也被认为是与疾病最相关的(这么说是因为前面提到过:一个模块里的基因是由表达模式相似的基因聚合而成的,大致就认为他们的功能相似)。
但实际上并不是模块中每个基因都跟性状相关,因此要进一步用MM和GS进行筛选。
MM(Module Membership): 是通过将某基因的表达量与模块特征基因——ME,进行相关性分析得到的,如果MM的绝对值接近1,说明基因与该模块的相关性很高,是该模块的重要组成部分,如果基因和某个模块的MM值为0,说明该基因与这个模块根本不相关,不属于这个模块。
GS(Gene Significance): 是基因和表型性状之间相关性的绝对值。具体来说,它是通过将指定基因的表达量与对应的表型数值进行相关性分析得到的相关性系数,GS越高,表明指定基因与研究表型越相关。
选择黄色模块并计算MM分数和GS分数
module <- c('yellow') probes <- colnames(datExpr) # 导入的fpkm文件中全部的基因名 inModule <- (moduleColors == module) # 模块是red中的基因 modProbes <- probes[inModule] # 筛选出关键基因 modGenes <- as.data.frame(modProbes) colnames(modGenes) <- 'modgene' ## 模块与基因间的相关性 geneModuleMembership <- as.data.frame(cor(datExpr, MEs, use = "p")) MMPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples)) geneTraitCor = as.data.frame(cor(datExpr, datTraits, use = "p")) geneTraitP = as.data.frame(corPvalueStudent(as.matrix(geneTraitCor), nSamples)) modNames = substring(colnames(MEs), 3) ## 获取关注的列 pheno = "disease" module_column = match(module, modNames) pheno_column = match(pheno, colnames(datTraits)) ## 获取模块内的基因 moduleGenes <- (moduleColors == module) MM <- abs(geneModuleMembership[moduleGenes, module_column]) GS <- abs(geneTraitCor[moduleGenes, 1]) 绘制MM和GS相关性散点图来评估模块中每一个基因与模块和性状的相关性。
verboseScatterplot(x = MM, y = GS, xlab = paste("Module Membership in", module, "module"), ylab = paste("Gene significance for", pheno), main = paste("Module membership vs. gene significance\n"), cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module) abline(h=0.6,v=0.8,col="blue",lwd=1.5) 结果如下图所示模块里的基因与模块的相关性系数为0.87,p值小于0.05,说明聚类很成功,模块里的基因与该模块相关。图中横坐标就是MM,纵坐标就是GS,图中蓝色的两条线就是MM > 0.8 & GS > 0.6的筛选条件。
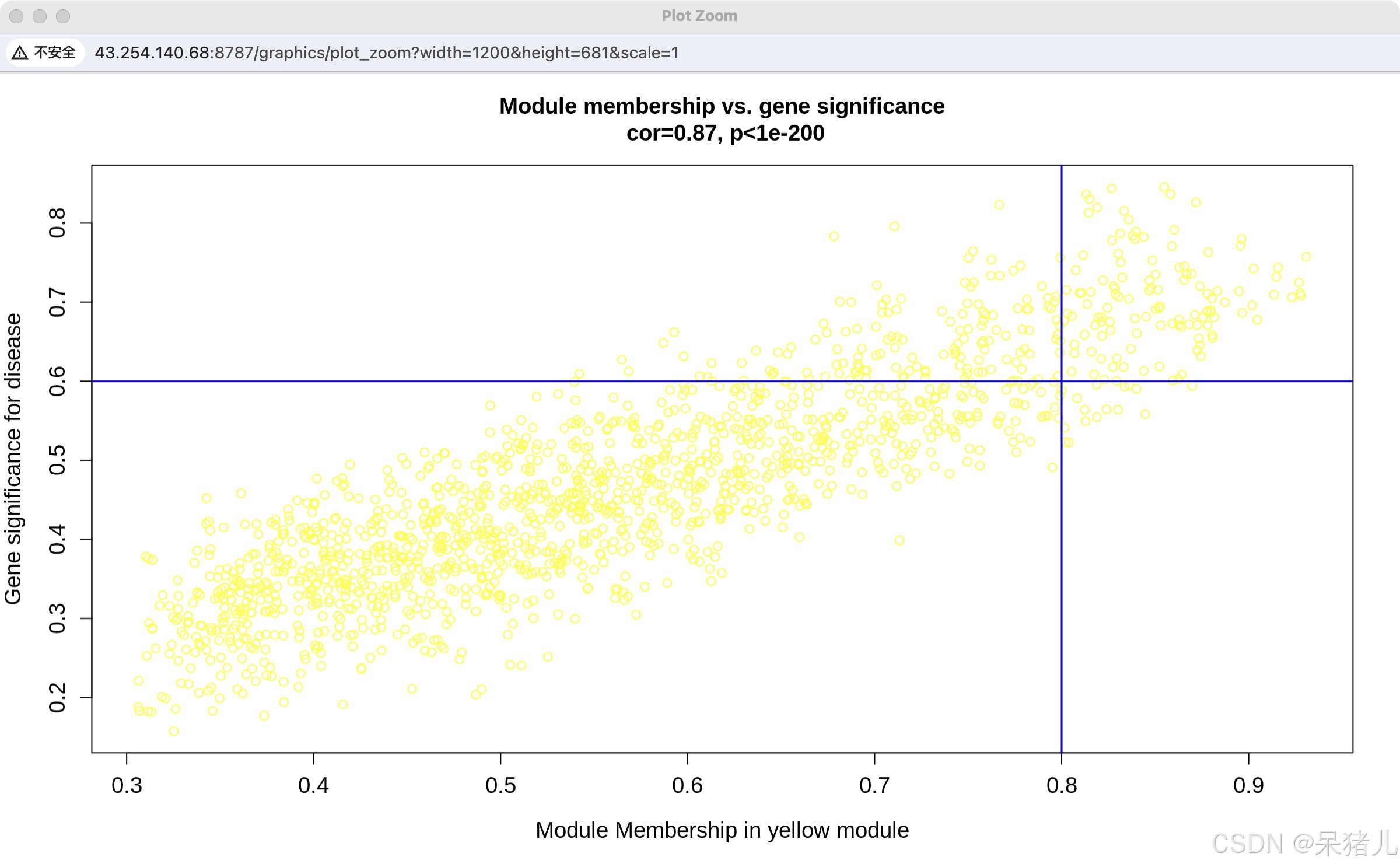
注:通常认为MM > 0.8 & GS > 0.6的基因不仅与模块高度相关,同时还与表型性状(肺腺癌)高度相关。
2. 9 提取通过筛选的基因作为关键基因(非必须)
前述提到MM > 0.8 & GS > 0.6的基因被认为更加重要,接下来就需要将这部分基因提取出来并保存进行后续分析。
c <- as.data.frame(cbind(MM, GS)) rownames(c) <- modGenes$modgene hub_gene <- subset(c, c$MM > 0.8 & c$GS > 0.6) write.csv(hub_gene, file = paste0(module, '_screen_gene_GS_MM.csv')) 这里面的hub_gene就是从黄色模块中挑选出来的更加重要的基因,这部分基因就被认为既与模块高度相关,同时也与表型性状高度相关,可进行后续分析。
注:最后这两部分写的非必须,是指可以不通过GS和MM筛选,直接拿模块里的全部基因去进行后续的分析,同时关于模块的选择也可以自己根据数据进行调整。
结语:
以上就是WGCNA的所有过程,如果有什么需要补充或不懂的地方,大家可以私聊我或者在下方评论。
如果觉得本教程对你有所帮助,点赞关注不迷路!!!
与教程配套的原始数据+代码+处理好的数据见配套资源
注:配套资源只要改个路径就能运行,本人已检测过可以跑通,请放心食用,食用过程遇到问题,可先自行百度,实在解决不了可以私信
- 目录部分跳转链接:零基础入门生信数据分析——导读
