
阅读量:0
摘要:近年来,获取RNA二级结构信息在RNA和基因功能研究中发挥了重要作用。虽然一些RNA二级结构可以通过实验获得,但在大多数情况下,仍然需要高效、准确的计算方法来预测RNA二级结构。目前的RNA二级结构预测方法主要基于最小自由能算法,通过迭代的方法找到RNA在体内的最佳折叠状态,以满足最小能量或其他约束。然而,由于生物环境的复杂性,真正的RNA结构始终保持生物势能状态的平衡,而不是满足最小能量的最佳折叠状态。对于短序列RNA,其RNA折叠生物体的平衡能量状态接近最小自由能状态;因此,最小自由能算法预测RNA二级结构具有更高的准确性。然而,在较长序列的RNA中,不断的折叠导致其生物势能量平衡偏离最小自由能状态。这种偏差是因为其结构复杂,导致其二级结构的预测精度严重下降。在本文中,我们提出了一种新颖的RNA二级结构预测算法,使用卷积神经网络模型结合动态规划方法来提高大规模RNA序列和结构数据的准确性。我们分析当前实验的RNA序列和结构数据来构建深度卷积网络模型,然后从大规模数据中提取有效分类的隐式特征来预测RNA序列中每个碱基的配对概率。对于获得的RNA序列碱基配对的概率,应用增强的动态规划方法来获得最佳的RNA二级结构。结果表明,我们提出的方法在预测三个基准 RNA 家族方面优于常见的 RNA 二级结构预测算法。根据深度学习算法的特点,可以推断,本文提出的方法与其他算法相比,预测成功率提高了30%,这是未来随着真实RNA结构数据量的增加所需要的。
1.介绍
RNA是生物体中重要的基本物质。它在基因的编码、解码、调节和表达方面发挥着重要作用。 RNA在生物体中的功能主要取决于其三级结构。然而,RNA分子的三级结构非常复杂,缺乏有效的表征方法;因此,从RNA分子的一级结构直接预测三级结构是非常困难的。因此,从RNA的一级结构预测RNA的二级结构成为研究RNA结构的主要过程。
目前,鉴定的RNA二级结构主要通过X射线衍射、核磁共振等生物实验手段获得。然而,在大规模测量结构时,生物实验方法效率低下、昂贵且艰巨(Novikova et al., 2012);此外,它们并非对所有 RNA 分子都有效 (Fürtig et al., 2003)。 Howard 和 Eran 提出了 PARS 技术来预测 RNA 二级结构 (Kertesz et al., 2010)。它应用核酸内切酶切割RNA的单链部分和双链部分,创建两个RNA片段文库,然后分别对两个RNA片段文库进行序列分析以获得RNA二级结构。但核酸内切酶不能穿过细胞膜,只能从细胞中提取RNA。这会破坏 RNA 的天然结构并导致结构变化。丁等人。 (2014) 使用 DMS 进行生物实验。 DMS可以与细胞中不配对RNA序列中的腺嘌呤和胞嘧啶反应,与DMS反应的RNA区域不能反转录成DNA。对逆转录成 RNA 的 DNA 进行序列分析以确定未配对的 RNA 区域。 DMS技术仍然存在缺陷。它只能确定RNA分子中的两个配对核苷酸,其余的需要计算机算法进行模拟。此外,研究人员使用SHAPE试剂代替DMS试剂(Wilkinson et al., 2008; Novikova et al., 2013),它可以酰化未配对状态下的四个碱基的2'羟基,从而分析单链RNA主链在任何位置的灵活性并推测碱基是否配对。但无法确定配对对象。到目前为止,还没有一种生物 RNA 方法能够大量预测真实的 RNA 二级结构;因此,仍然需要计算预测算法来有效预测RNA二级结构。
主流的RNA二级结构预测算法主要有两类。一是确定性动态规划算法。最早使用的动态规划算法是基于最大碱基配对数的 Nussinov 算法(Nussinov et al., 1978)。该算法简单地假设RNA单链折叠成自身,以便碱基对可以(尽可能多地)构成RNA的二级结构。但该算法由于假设前提过于简单,且形成的碱基对往往不连续,无法形成茎区,预测精度较低。基于Nussinov算法和能量信息,Zuker提出了最小自由能算法(Zuker and Stiegler,1981)。最小自由能算法假设RNA结构与自由能有很大关系。自由能的大小不仅与碱基配对的类型有关,而且自由能的大小还受到相邻碱基对的影响。不同结构(毛环、内环等)的自由能也有很大差异。最小自由能算法仍然采用动态规划的思想,但计算对象是通过实验获得的一系列复杂的自由能参数。许多著名的RNA二级结构预测软件应用,例如mfold网络服务器(Zuker,2003)和RNAfold(Hofacker等,1994),都采用了最小免费算法及其改进。然而实验表明,由于内部环境的复杂性,RNA很少以能够最小化结构自由能的方式折叠,一般处于次优能量折叠结构(Zou et al., 2008)。值得注意的是,Zuker 算法对于较短 RNA 的二级结构具有更好的预测结果。然而,对于较长的 RNA,其预测准确性急剧下降。
第二类主流RNA二级结构预测算法是指比较序列分析方法。在生物实验中,通常需要同时处理一组或多组同源RNA序列。一般认为,同源RNA分子中,结构的保守性大于序列的保守性。例如,所有 tRNA 分子的二级结构都是三叶草形状的。这种形状的一致性使 tRNA 分子具有执行类似功能所需的结构一致性。因此,比较序列方法可以在一定程度上提高预测精度。比较序列分析的方法主要有3种。第一种方法包括 RNA 结构的先验分布,其中包括比较和事后预测时的进化历史(Knudsen 和 Hein,1999)。该方法获得的结果很大程度上取决于多序列比对的效果。第二种方法同时进行结构预测和序列比较,但该算法消耗过多的计算资源(Sankoff,1985)。第三种比较序列分析方法是先预测,后比较。该方法可以获得多个候选结构,但不能保证包含真实结构(Allali and Sagot,2005)。
人工智能方法已在许多领域得到应用。目前已经出现了一些人工智能学习算法如遗传算法(Hu, 2003)、神经网络算法(Zhang et al., 2006)、支持向量机算法等方法来预测RNA的二级结构。均取得了良好的成绩。然而,这些方法都是基于小样本,对于单类数据样本的预测精度较低。随着计算机技术的发展,人工智能领域出现了深度学习方法,可以有效提高预测的准确性。深度学习方法可以通过大规模数据中的深层网络提取有效的隐式特征,并利用这些特征构建有效的预测模型。目前,深度学习方法在蛋白质二级结构预测领域取得了巨大突破(Wang et al., 2016)。然而,与蛋白质二级结构预测相比,RNA二级结构预测更加复杂和困难,因为即使蛋白质的每个氨基酸与其他氨基酸不相关,RNA上的每一对碱基都需要对应于链中的另一个碱基结构预测期间链中的酸。本文提出了一种将深度学习与动态规划相结合的RNA二级结构预测计算方法,可以有效解决上述问题。与当前主流算法相比,我们的方法具有更好的结果。
2.数据和方法
RNA二级结构主要由连续碱基互补配对形成的茎结构和碱基不配对形成的环状结构组成。这种RNA二级结构也称为茎环结构,只要确定了RNA序列的所有配对碱基,就可以确定整个RNA的二级结构。基于迄今为止我们文献检索中提出的RNA二级结构预测问题,本文提出了一种更有效的RNA二级结构预测算法。该算法称为 CDPfold,结合了卷积神经网络和动态规划以及序列比对方法。在比较序列分析中,我们构建了一个卷积神经网络,从大规模数据中提取有效隐式特征的特征,并预测每个碱基在RNA序列上的匹配概率。卷积神经网络可以使用当前收集的RNA序列作为训练样本,解决了比较序列分析中同源序列的限制。对于卷积神经网络得到的概率结果,我们利用动态规划的迭代思想和RNA二级结构的定义,得到碱基匹配概率和最大RNA二级结构。该操作可以避免由于使用自由能方法而导致的长序列预测精度的下降。 CDPfold预测RNA二级结构的过程如图1所示。
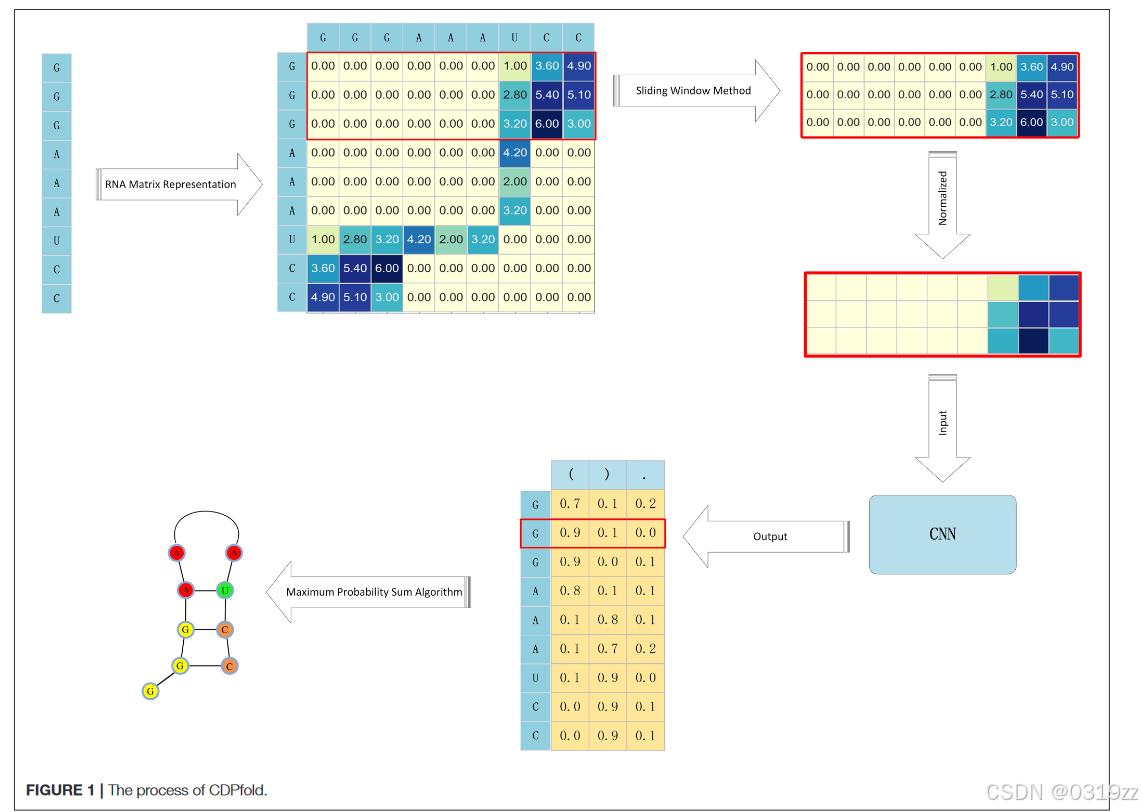
2. 1基于RNA序列配对的RNA矩阵表示
RNA序列主要由“A”、“U”、“G”、“C”四种碱基组合组成,但大多数算法模型不接受“AUGC”组合序列作为输入数据。因此,我们必须对序列进行编码。目前最常见的编码方法是one-hot编码,但由于one-hot编码不能体现碱基之间的隐式匹配,因此我们开发了一种新的编码方法。
我们为每个 RNA 构建了矩阵 Wi×i,其中矩阵的每一行代表该位置可能的碱基配对,如下所示:
1.根据配对碱基之间的氢键数量,将A和U之间的配对权重设置为2,将G和C之间的配对权重设置为3。由于U-G对是摆动碱基对,因此U 和 G 之间的配对权重设置为 x (0 < x < 2),这导致:
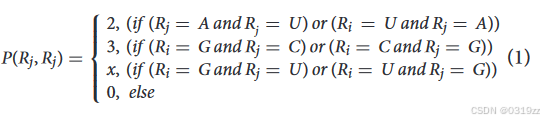
2. 对于任意两个位置,比如RNA序列上的i、j,本文不仅要考虑这两个碱基的配对,还要考虑这两个位置能否在茎上形成配对的碱基。因此,我们必须考虑 i 左(右)侧碱基与 j 右(左)侧碱基的配对。
3.对于一根茎来说,茎中部的配对碱基相对稳定,两侧的配对碱基相对不稳定。因此,该计算提供了将 RNA 序列上的两个位置 i 和 j 配对的可能性。本文参考了局部加权线性回归的思想,加入了高斯函数作为权重。 i 和 j 越接近,配对碱基的权重越高,其效果也越大。
结合这些观点,本文引入以下算法流程来计算编码矩阵Wi×i各位置的具体值,如图2所示。
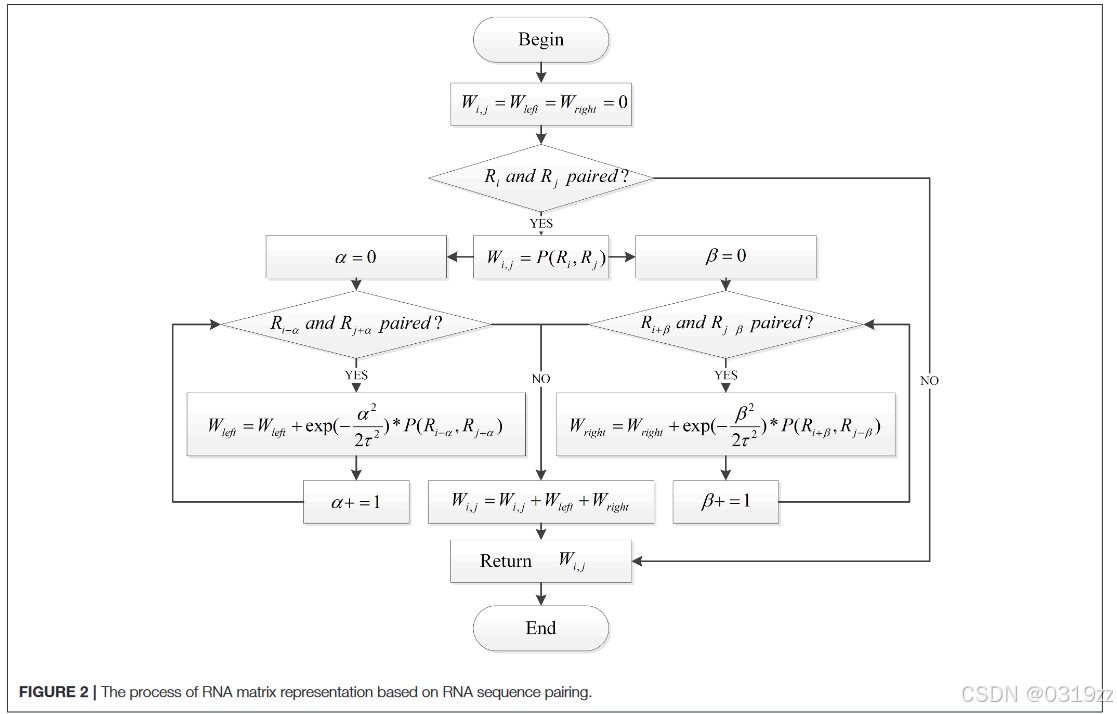
根据RNA序列编码矩阵的碱基配对可以通过计算得到。通过对矩阵的分析,我们可以知道,茎区在RNA真实结构中的位置是用编码矩阵中一条中间值大、两边值小的次对角线来表示的。深度学习方法中卷积神经网络的优势在于可以有效地提取矩阵中块的区域特征。因此,我们使用卷积神经网络而不是其他机器学习模型来预测 RNA 序列中碱基的配对。
2.2卷积神经网络预测RNA序列碱基配对的概率
我们的目标是预测 RNA 序列上每个碱基的配对;因此,我们必须拆分 RNA 序列编码矩阵。 RNA表示方法将长度为n的序列转换为大小为n×n的矩阵。我们使用滑动窗口方法将矩阵划分为n个大小为d×n的矩阵。其中d是滑动窗口的大小。因此,每个 RNA 序列上的碱基可以用大小为 d × n 的矩阵表示。滑动窗口的大小,采用滑动窗口方法,对实验影响很大。如果滑动窗口设置得太小,提取的特征将不完整。太大的窗口设置会导致矩阵中冗余信息较多,从而导致训练模型时间较长,并可能影响最终预测模型预测的准确性。经过分析,滑动窗口的值应该与RNA中茎区的长度有关。因此,我们必须统计实验对象的茎区域信息来确定滑动窗口的大小。
卷积神经网络要求输入模型的数据大小统一,而每个RNA序列对应的RNA序列由于RNA序列的长度不同,其大小也不同。因此,在实验过程中,我们需要计算实验数据集中RNA序列长度的平均值,并使用该平均值对数据进行归一化。滑动窗口方法和RNA编码矩阵的归一化可以将长度为n的RNA序列转换为n个相同大小的矩阵,满足卷积神经网络对输入数据的要求。
本文使用点括号表示法来表示RNA二级结构。点括号表示RNA二级结构表示为“(“,”)”和“.”序列的组合。因此,本文设计的卷积神经网络的输出层由三个节点组成,每个base的输出对应于三个标签“(”,“)”和“.”的概率对应的矩阵。
2.3最大概率和算法纠正预测
深度学习方法对于分类问题的准确率很高。然而,RNA二级结构预测并不是一个简单的分类问题。我们可以将RNA二级结构预测视为在一定限制下的多个分类问题的组合。
根据上一步的结果,我们可以得到Pleft、Pright和Ppoint。这是三个标签“(“,”)”和“.”的概率。 RNA序列中每个碱基的二级结构。然而,如果使用预测概率最高的标签作为每个碱基的预测结果,这种组合并不能保证这样的结果满足为RNA定义的二级结构的定义:可能会出现左侧的数量括号不等于右括号的数量,或者可以预测匹配的括号不能与相应的碱基配对。因此我们需要修改预测结果以满足RNA二级结构定义的要求。
基于卷积神经网络上一步获得的概率结果,本文的目标是找到一个兼容的括号序列来表示RNA的二级结构。为了实现这一目标,该过程需要:
1.序列括号匹配。
2.匹配序列中的括号,将相应位置的碱基配对(A-U、G-C、G-U)。
3.为了满足 1 和 2,根据其卷积神经网络内每个标签的输出来最大化概率的总和。
为了找到满足这些要求的序列,本文增强了动态规划方法中的Nussinov算法。这就需要将Nussinov算法中迭代累加的配对碱基数改为迭代累加碱基的累加概率之和。因此,提出了最大概率和算法。该算法利用了动态规划方法。通过多次迭代,可以获得满足要求的RNA二级结构。具体迭代公式如下:
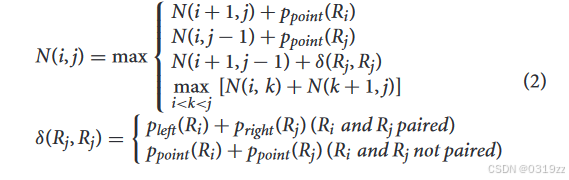
其中N(i,j)是RNA序列中第i个碱基到第j个碱基的最大概率。 Pleft、Pright 和 Ppoint 表示对于三个标签,卷积神经网络输出 RNA 序列的第 i 个碱基的概率。
3.结果
3.1通过 CDPfold 预测单家族 RNA 的二级结构
我们实验中使用的数据来自 Turner 和 Mathews (2009)。数据集中包含的数据如表1所示。

在数据集中包含的各个RNA家族中,我们首先选择数量最多且分布最集中且没有假结的5sRNA。对5sRNA数据集的序列分析表明,RNA数据集中存在一些相同或相似的序列数据。为了避免相同或相似的序列数据对实验产生影响,需要对数据集中的数据进行预处理。即5sRNA数据集被编程以去除数据中相同或相似的序列。去重操作后,实验中使用的5s RNA数量为1,059。为了训练模型并准确评估整个模型,我们将删除的 5sRNA 数据集的数量划分为训练集,由验证集和测试集组成。训练集、验证集和测试集的RNA比例为7:2:1。
实验使用训练集训练网络模型并确定模型参数;然后,使用验证集进行模型选择。因此,在模型的最终优化和确定中,最终的测试集是用来衡量整个预测方法的泛化能力的。
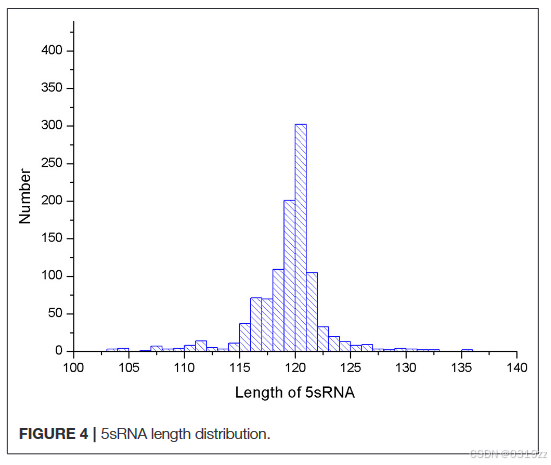
CDPfold中的几个参数会影响实验结果,有问题的参数必须在实验前修复。第一个问题参数是滑动窗口的大小。我们计算了实验中使用的5sRNA数据集中所有RNA的最大茎区的长度。得到的结果如图3所示。使用5sRNA数据集中最长茎区域的长度作为滑动窗口方法的大小。我们还计算了数据集中5sRNA序列的平均长度,如图4所示。
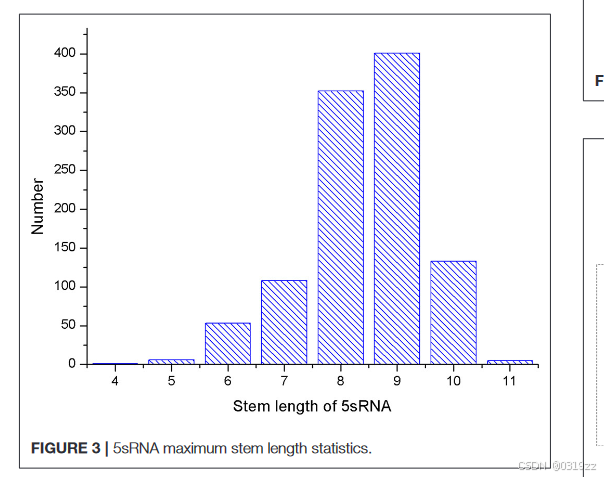
图4显示5sRNA的最大茎长度为11个连续碱基对,序列平均长度为120 nt。由于卷积神经网络对于平移和缩放后的图像具有良好的精度,因此本文应用了图像缩放的思想,这意味着通过滑动窗口获得的基数的矩阵表示可以均匀缩放为11×的矩阵大小120.
本文构建的卷积神经网络模型使用的框架是Tensorflow。卷积神经网络模型由一个输入层、三个卷积层、三个池化层、两个全连接层和一个最终输出层组成。在测试阶段,去掉输出层的tf.nn.top_k()函数,得到每个base对应三个标签的概率。本文使用的卷积神经网络模型如图5所示。从卷积神经网络输入层输入的数据通过滑动窗口算法和归一化基矩阵来表示。使用批量随机梯度下降的参数优化方法每次迭代使用 256 个数据。卷积神经网络由三个卷积层、三个池化层和两个全连接层组成,其中每个卷积层使用16个3×3卷积核,每个池化层也使用3×3卷积核。最大的池,每个全连接层使用32个节点。模型的输出层将数据映射到点括号表示的三个标签上,可以验证基点属于三个标签的概率。模型各参数的初始化方法为Xavier初始化方法,输出层的误差函数采用最大熵函数。当模型参数训练完成后,模型迭代400次。
通过滑动窗口方法和归一化,可以获得训练集中5s RNA序列中每个碱基对应的矩阵表示,其中每个碱基都有对应的结构标签。分析数据可知,由于每个5s RNA序列中未配对碱基的数量略大于配对碱基对的数量,这会导致数据集中三类数据样本的不平衡,因此数据需要对不平衡数据进行处理。由于实验数据量充足,因此将采用上采样数据处理方法来平衡数据集中的各个样本数据。
处理后的数据用于训练卷积神经网络模型。我们在训练集和测试集上构建的卷积神经网络模型的性能如图6所示。从图6中我们可以看出,该模型在训练集和测试集上的测试精度相似,实验结果表明:结果没有过度拟合。该图也表明模型在训练集和测试集上的测试精度相似,实验结果没有过拟合。

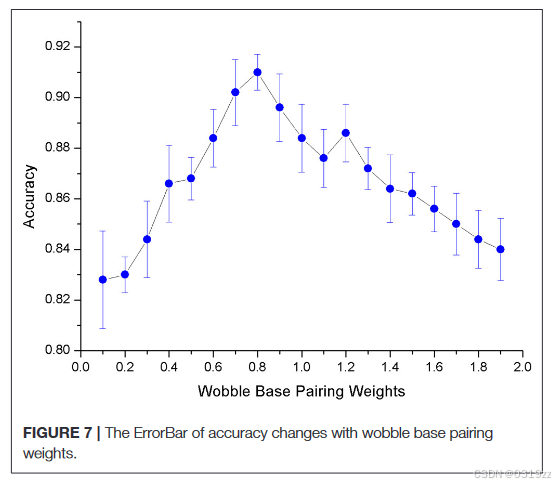
在确定实验中使用的模型后,我们需要为 G-U 配对的权重 x 选择一个合适的值(公式 1)。摆动配对的匹配权重不宜过大或过小。不利的权重会导致预测准确率下降。为了选择合适的权重,我们进行了多次实验。实验结果如图 7 所示。实验结果表明,当 G-U 配对的匹配权重为 0.8 时,整个模型的准确率均值和方差都是最佳的。
测试集数据被输入训练有素的 CDPfold,卷积神经网络获得的每个 RNA 上每个碱基的配对概率被用作中间结果。这些中间结果用于我们的概率和最大校正算法。这样就得到了符合 RNA 二级结构定义的最优二级结构,并与相应的真实结构进行了比较,从而验证了我们的完整模型设计。
对于 CDPfold 所获得的 RNA 二级结构的预测,我们使用了灵敏度和特异性两个指标。灵敏度指的是真实结构中所有碱基对的预测百分比,相当于机器学习中的召回率。特异性是指所有预测碱基对的正确率,相当于机器学习中的精确率。RNA 二级结构预测算法在一般情况下很难实现,因为它总是偏向一边。F 分数可用于衡量精确率和召回率。
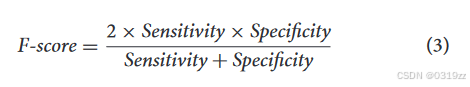
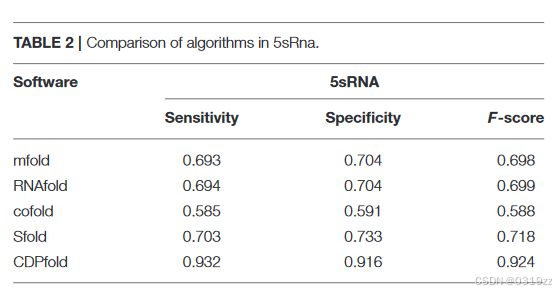
根据上述指标,我们得到了所设计算法模型在 5sRNA 数据集上的预测效果。我们使用相同的数据在其他已发布的算法下进行了实验。表 2 比较了我们的新算法与当前软件中其他流行程序的实验结果。表 2 显示了我们设计的算法与其他算法在 5sRNA 数据集上的准确性比较。很明显,我们设计的算法的灵敏度和特异性明显高于其他算法。
3.2用 CDPfold 预测多家族 RNA 的二级结构
基于上述研究,我们使用 5sRNA 数据集训练的模型来预测 tRNA 的二级结构。结果灵敏度为 0.2,特异度为 0.15,这与 5sRNA 数据集训练模型在 5sRNA 数据集上的效果大相径庭。我们对这一结果进行了分析,发现 5sRNA 的功能与 tRNA 的功能截然不同。使用 5sRNA 数据集训练的模型只提取有利于 5sRNA 分类的特征。tRNA 缺乏这些特征,导致预测准确率大大降低。因此,在未确定 RNA 功能或家族的情况下,无法使用已建立的模型直接预测这些特征。因此,必须获得整个样本数据才能建立通用模型。
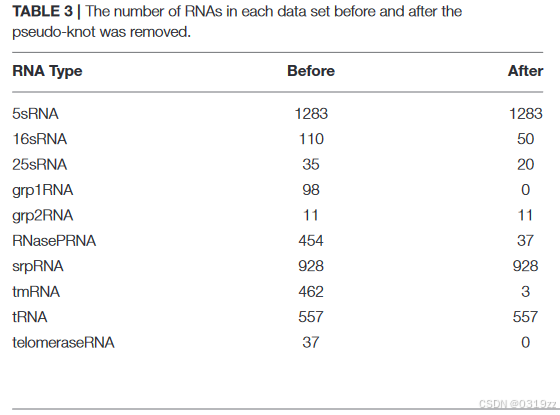
首先,我们分析了数据集中的所有数据,在一些 RNA 结构数据中发现了伪节点。由于假结属于 RNA 三级结构范畴,因此在预处理操作中删除了所有带有假结的数据。表 3 显示了删除伪节点后的 RNA 数量。
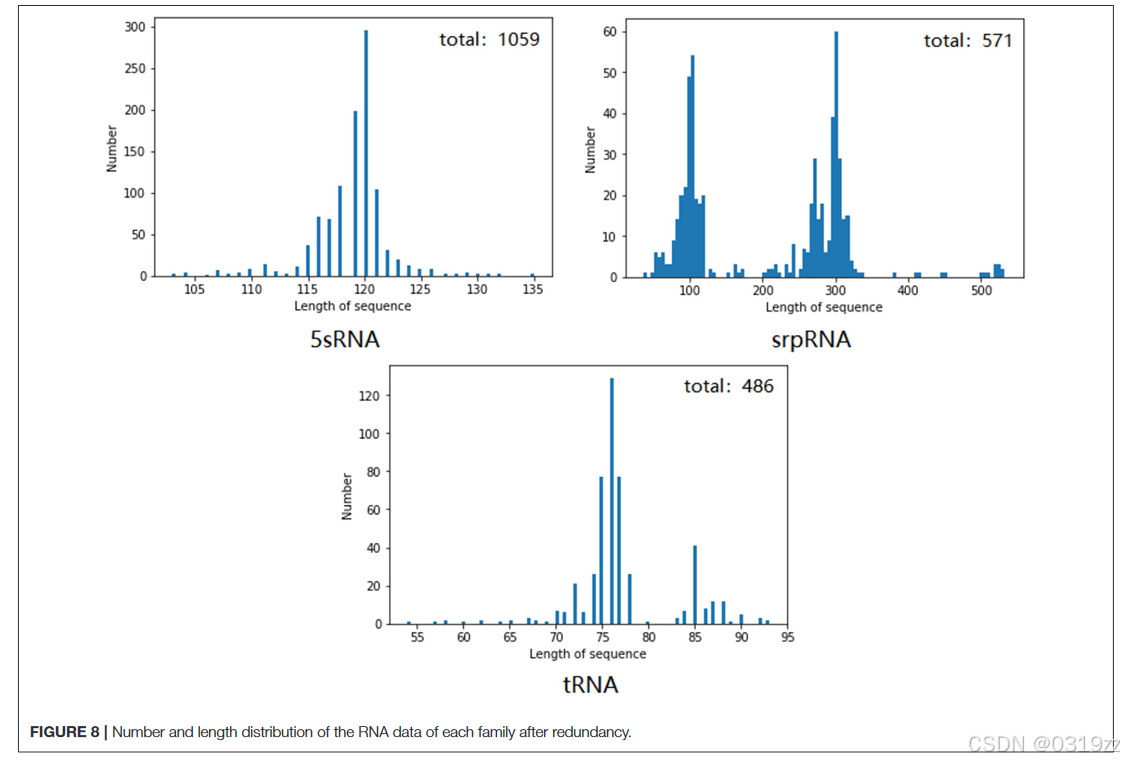
我们选择了假结点后数字大于 100 的 5sRNA、srpRNA 和 tRNA,首先对这三类 RNA 进行了去除冗余操作,删除了 RNA 数据集中相同或相似的序列数据。图 8 显示了去除相同或相似序列数据后的各族 RNA 和数据分布情况。
我们还计算了数据集中每个 RNA 序列的最大茎干长度和 RNA 序列的平均长度,如图 9 和图 10 所示。
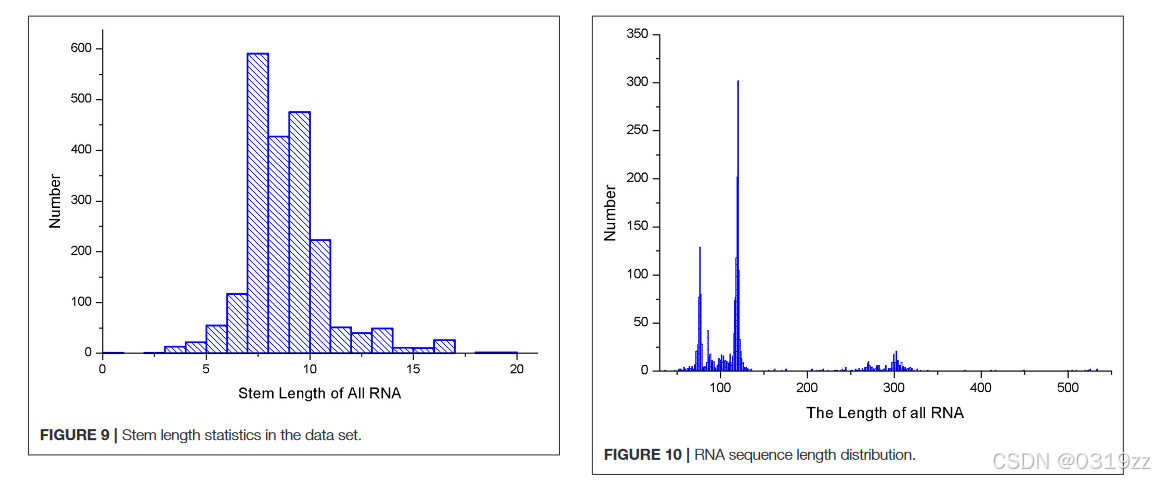
从图 9 和图 10 中可以看出,茎区的最大长度为 19 个连续碱基对,平均长度为 128 nt。因此,经过归一化和滑动窗口操作后,RNA 矩阵表示为大小为 19 × 128 的矩阵。实验将数据分为训练集、验证集和测试集,其中各类 RNA 的比例保持为 7:2:1。在一般模型中,由于数据类型和 RNA 数量大幅增加,实验将对原始 5sRNA 预测模型的卷积神经网络进行微调:为了提取各种 RNA 的更通用的隐藏特征,卷积层和池化层的数量从三层减少到两层。其他配置参数没有变化。在训练过程中,每次迭代的批量数据大小从 256 增加到 512,迭代次数增加到 2,000 次。图 11 中,实验中使用的卷积神经网络模型如下。
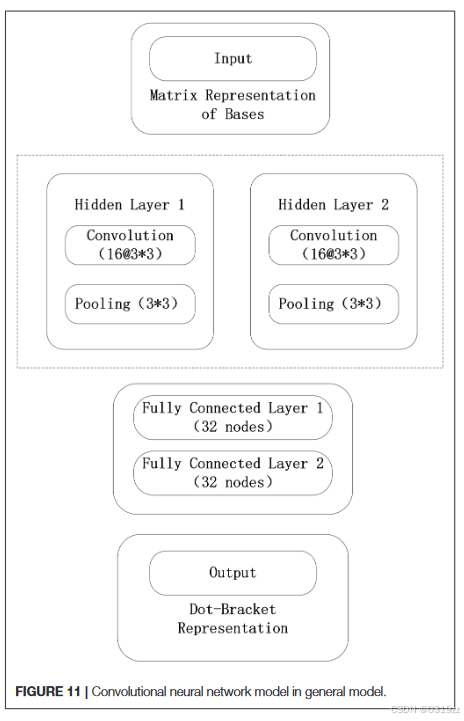
我们的卷积神经网络模型训练完成后,将测试集数据输入训练好的通用模型,以获得每个 RNA 上每个碱基的配对概率,并使用最大概率和碱基校正算法对配对概率和 RNA 序列进行校正。通过这种方法,可以得到 RNA 序列的最佳二级结构。
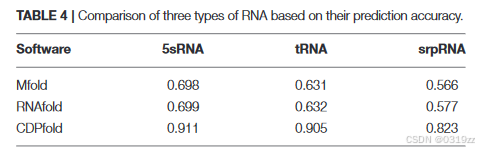
利用 F 分数,我们可以得到所设计的通用模型在三种 RNA 数据集上的预测效果。我们使用相同的测试数据在其他已发布的算法下进行实验。比较结果如表 4 所示。
4.讨论
本文提出了一种基于卷积神经网络的RNA二级结构CDPfold预测方法。该方法利用卷积神经网络提取 RNA 序列数据的隐藏特征,并将其应用于结构预测领域。利用基于动态编程的修正算法对结果进行修正,以获得最佳的 RNA 二级结构。实验证明,我们的方法在预测 RNA 二级结构的准确性方面表现良好。
虽然 CDPfold 在 RNA 二级结构预测方面取得了不错的成果,但在实验过程中也遇到了一些问题,现总结如下,并提出解决问题的建议。
首先,RNA 之所以能形成茎区,取决于碱基互补配对形成的氢键。DNA 的二级结构主要以双螺旋的形式存在。由于双螺旋结构的限制,DNA 中的碱基配对只能由嘧啶和肼对组成。因此,DNA 分子只有 A-T 和 G-C 两种配对方式:而 RNA 分子则不同。RNA 分子主要以单链形式存在。它们的双链区域由同一链上的不同区域组成。它们不具有长结构的规则双螺旋结构。因此,除了标准的 A-U 和 G-C 碱基对外,还有 G-U 摇摆对。摇摆对形成的氢键不稳定,并非所有的 G 和 U 元素都能形成成对碱基对。目前对GU摆动配对的处理方式将被固定下来,要么是可配对,要么是不可配对。本文选择了一个较小的数字作为 G-U 摆动配对的配对权重,但 G-U 配对问题并没有得到很好的解释。我们认为有必要根据 RNA 折叠过程中的不同状态来动态判断 G 和 U 能否形成配对碱基对,但这种动态方法难度极大,目前还没有研究提出相应的解决方案。
本文提出的 CDPfold 方法预测的结果仍需进一步修正卷积神经网络预测的结果。这是因为所有机器学习算法都存在泛化误差,而卷积神经正是由于泛化误差的存在而导致的。网络得到的结果并没有形成令人满意的 RNA 二级结构。在其他使用机器学习算法解决 RNA 二级结构预测的研究中也出现了类似的情况。要获得令人满意的 RNA 二级结构以进行更准确的预测,主要有两种解决方案。一种是直接优化机器学习模型的结果。本文采用的就是这种方法。第二种是将结果作为条件约束,并利用这些约束来优化其他算法。从本质上讲,这两种方法都是对中间结果的优化过程。在这个问题中,生成反网络模型可能是一个有效的解决方案。生成反网络的生成器用于生成 RNA 二级结构,判别器用于确定结果是否满足 RNA 二级结构的定义。通过生成器和判别器之间的对抗,可以获得最佳的 RNA 二级结构。这种方法的难点在于如何设计一种好的训练方法。否则,由于生成模型的自由性,输出结果可能不尽人意。
在优化算法的选择方法上,本文作者采用了遗传算法等群体智能优化算法。这些智能算法可以通过模拟生物进化来解决复杂的非线性问题。本文将卷积神经网络提供的概率结果作为遗传算法中的选择、突变、杂交等概率,将模拟 RNA 结构物种中的错配数作为优化目标。虽然这种方法也能得到符合要求的 RNA 二级结构,但由于群智能优化算法中每个环节的随机性和数据的离散性,使得算法无法有固定的优化迭代次数。此外,由于本文的目标是找到不失配的 RNA 二级结构,而这种结果的数量很大,每次优化的结果都具有不确定性,因此不能采用群体智能算法作为本文的优化算法。因此,本文选择动态编程算法作为优化算法,并在努西诺夫算法的基础上提出了概率法和最大修正法。
在目前的 RNA 二级结构预测中,假节的预测仍是一个难点。本研究发现,5sRNA、srpRNA 和 tRNA 不存在假节,而大多数 RNasePRNA 和 tmRNA 存在假节。在这些含有假节的 RNA 中,每条 RNA 中的假节数量相对较少,但它们的存在不容忽视。假结不仅对 RNA 的功能起着重要作用,而且假结效应的预测错误,会造成正常干区的错误。本文采用的 RNA 结构表示方法是点括号表示法。然而,点括号表示法并不能反映 RNA 结构中存在的假结。因此,在实验中删除了包含假结的数据。如果能找到一种能表示假结的 RNA 二级结构表示法,本文提出的 CDPfold 可以进行相应的修改,以预测带有假结的 RNA 二级结构。
本文使用的实验数据主要集中在 5sRNA、srpRNA 和 tRNA 上。这三类 RNA 序列的长度大多在 50 到 350 nt 之间。在这部分长度范围内,CDPfold 的效果是由现有 RNA 预测软件造成的。而对较长的 RNA 序列的二级结构预测则没有体现出来。这是因为目前的实验方法还不够完善。通过实验测得的长序列 RNA 二级结构数据还不够多。实验中使用的数据集由 Turner 和 Mathews(2009 年)提供。使用的长度超过 1,000 nt 的 RNA 序列不到 200 个,占整个数据集的 10%以下。影响深度学习预测效果的最重要因素是数据量,因此我们没有研究更长的序列。不过,随着实验技术的不断改进,实验所测得的长序列结构数量在不断增加。在此基础上,基于深度学习的模型具有优势。
最后一点是 RNA 结构的不稳定性。RNA 分子的结构极易受到环境因素的影响。研究表明,RNA 分子在体外环境中会破坏其天然结构,导致结构损伤;因此,体内结构预测实验并不完美,这意味着当前的 RNA 二级结构并不一定是真实结构。
此外,与蛋白质的功能不同,并非所有的 RNA 分子都能在体内发挥作用;而且,编码蛋白质的 RNA 只占 RNA 总量的 2%。因此,不具备实际功能的 RNA 结构可能不如功能性 RNA 结构那样固定。这些问题都会对 RNA 二级结构预测产生影响。
总的来说,基于卷积神经网络的CDP-Fold算法用于RNA二级结构预测,在无伪点的数据集上取得了良好的结果。在 RNA 二级结构预测的研究中还存在很多困难,很多地方还需要改进。我们的研究为 RNA 二级结构的研究提供了新的思路,也为其他研究人员提供了很好的结构预测问题和解决方案。
